Noonan综合征:症状体征、病因、流行病学、诊断和治疗
2022-08-17 MedSci原创 MedSci原创

Noonan综合征是一种先天性疾病,大多数病例为散发性,家族性患者为常染色体显性遗传,基因突变是基本的病因。国外报道发病率为1/2500~1/1000活产儿。特征性表现包括特殊面容、身材矮小、胸廓畸形
Noonan综合征是一种先天性疾病,大多数病例为散发性,家族性患者为常染色体显性遗传,基因突变是基本的病因。国外报道发病率为1/2500~1/1000活产儿。特征性表现包括特殊面容、身材矮小、胸廓畸形、先天性心脏病和凝血障碍等。尚无根治方法,只能通过定期随访及时发现异常,并予以对症治疗。预后主要与心脏病变的严重程度有关。2018年5月11日,该疾病被列入国家卫生健康委员会等5部门联合制定的《第一批罕见病目录》。
一、一般概述
Noonan 综合征是一种遗传性疾病,通常在出生时就很明显(先天性)。这种疾病的特点是症状和身体特征范围广泛,范围和严重程度差异很大。在许多受影响的个体中,相关的异常包括独特的面部外观;宽颈或蹼颈;后发际线低;典型的胸部畸形和身材矮小。头部和面部(颅面)区域的特征性特征可能包括广泛的眼睛(眼距过远);可能覆盖眼睛内角的皮肤褶皱(内眦褶皱);上眼睑下垂(上睑下垂);小颚(小颌);鼻根凹陷;鼻子短,基部宽;和低位,向后旋转的耳朵(耳廓)。通常还存在明显的骨骼畸形,例如胸骨异常(胸骨)、脊柱弯曲(后凸和/或脊柱侧凸)和肘部向外偏移(肘外翻)。许多患有 Noonan 综合征的婴儿也有心脏(心脏)缺陷,例如从心脏右下腔到肺部的正常血流受阻(肺动脉瓣狭窄)和心室心肌增厚(肥厚型心肌病)。其他异常可能包括某些血液和淋巴管的畸形、凝血和血小板缺乏、学习困难或轻度智力障碍、受影响的男性在出生后第一年睾丸未能进入阴囊(隐睾),和/或其他症状和发现。
在大多数情况下,Noonan 综合征是一种常染色体显性遗传疾病,由超过 8 个基因的异常(突变)引起。五个最常涉及的基因是:PTPN11 (50%)、SOS1 (10-13%)、RAF1 (5%)、RIT1 (5%) 和 KRAS (小于 5%)。 NRAS、BRAF、MEK2、RRAS、RASA2、A2ML1 和 SOS2 突变的个体较少。发现与 SHOC2 和 CBL 突变有关的 Noonan 样疾病。由 LZTR1 致病变异引起的 Noonan 综合征可以以常染色体显性或常染色体隐性方式遗传。
二、症状与体征
Noonan 综合征患者的相关症状和身体检查结果因人而异,其范围和严重程度差异很大。一些受影响的人只有轻微的面部异常;其他人可能有与该疾病相关的大部分症状和发现,例如头部和面部(颅面)区域的独特特征,宽颈或蹼状颈部,身材矮小,骨骼畸形,先天性心脏缺陷,某些血液和淋巴畸形血管、凝血和血小板缺乏、注意力问题、轻度智力障碍和/或其他异常。
大多数患有 Noonan 综合征的婴儿具有特征性颅面特征。在许多情况下,头部显得比较大。受影响的婴儿可能有几种影响眼睛的发现,包括异常突出的广泛分布的眼睛(眼距过远);上眼睑下垂(上睑下垂)和/或异常厚的“蒙面”眼睑;内翻或外翻的眼睛(斜视);向下倾斜的眼睑(睑裂);可能覆盖眼睛内角的皮肤褶皱(内眦赘皮);和/或眼睛的明显蓝色或蓝绿色部分(虹膜)。
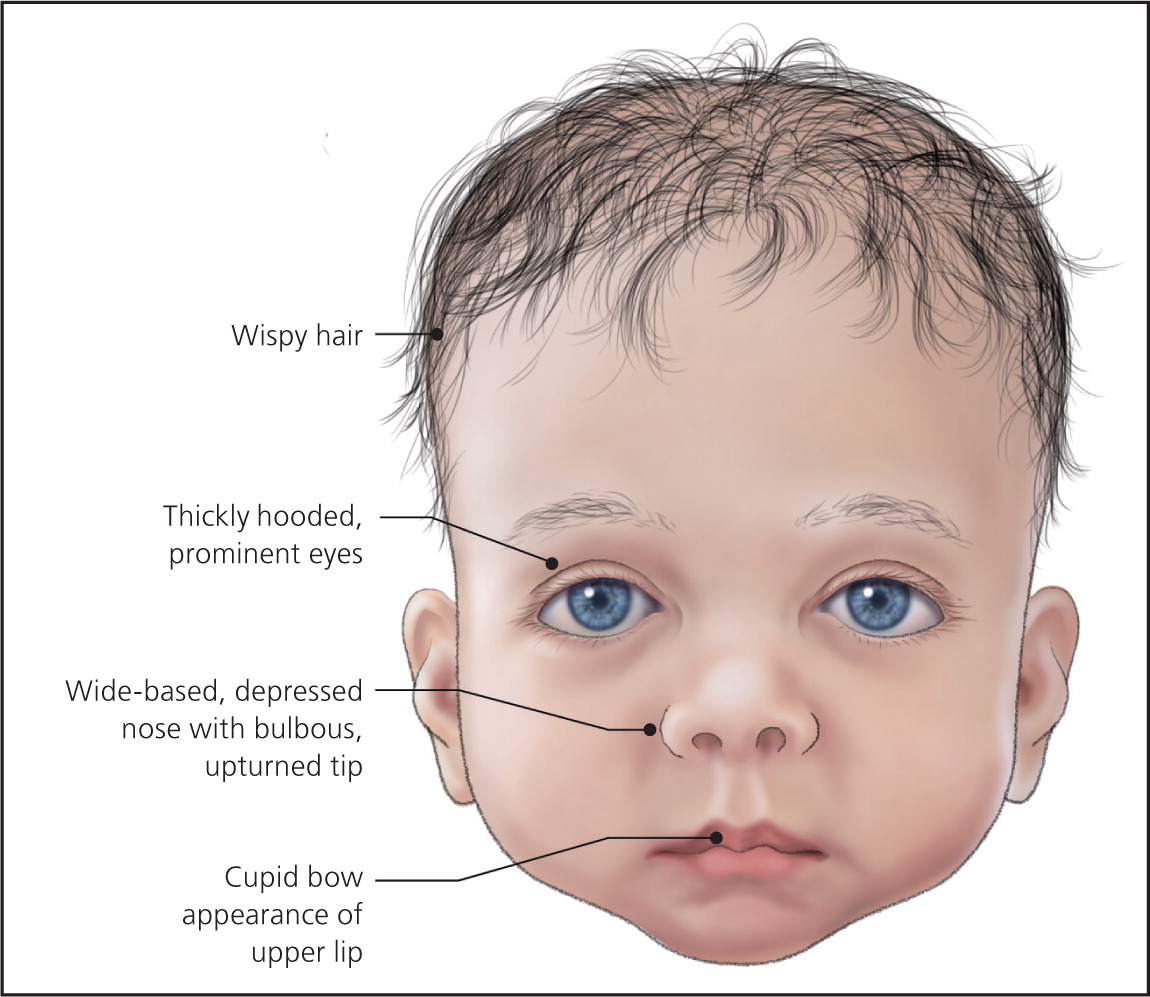
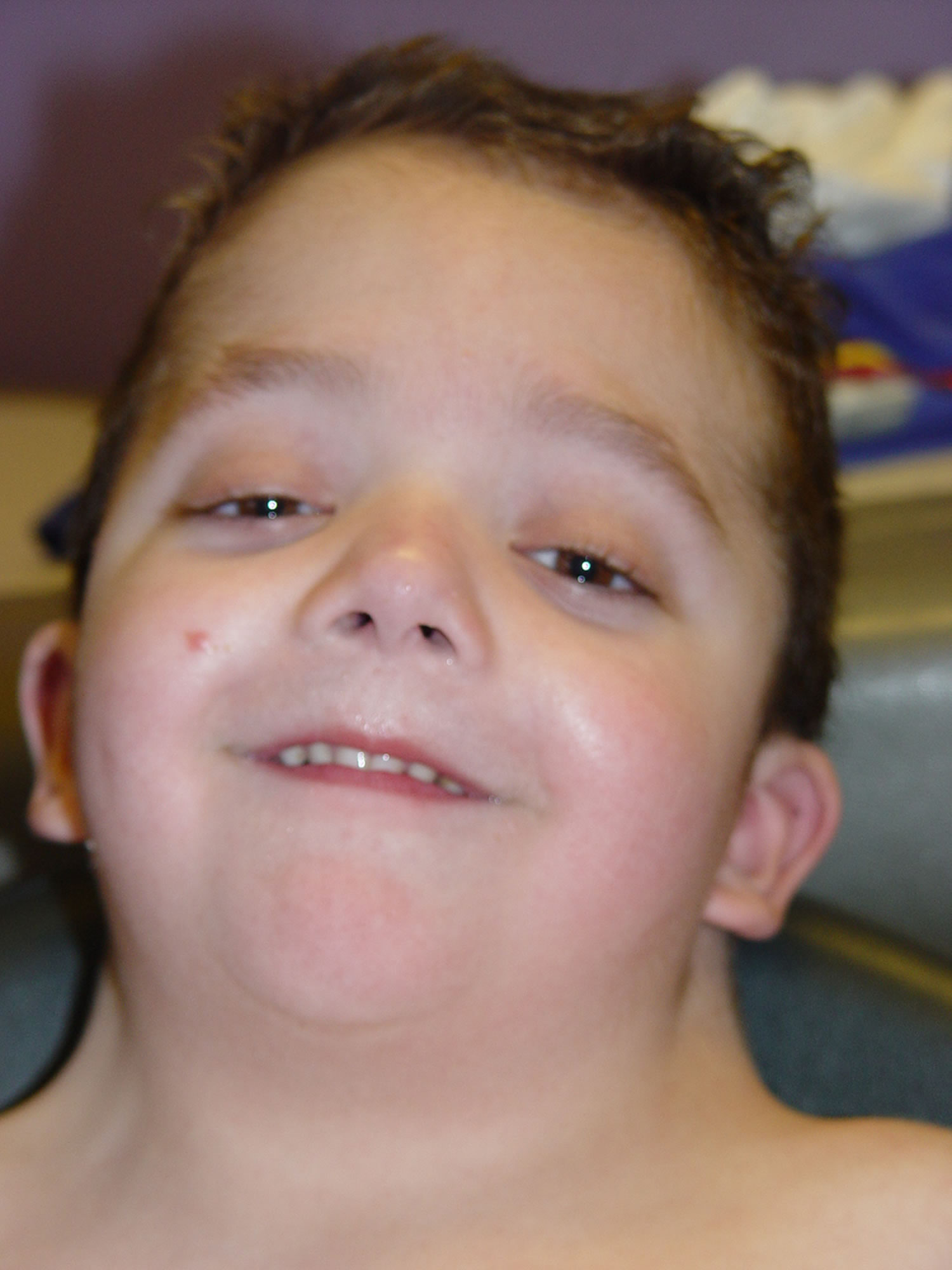
许多患有 Noonan 综合征的婴儿还具有额外的颅面特征。这些可能包括上唇(人中)中部异常深的垂直凹槽;和/或小下巴。受影响的婴儿也可能有小颌(小颌畸形);下齿拥挤,低位,后旋外耳(耳廓);和/或鼻子的明显异常,包括鼻根凹陷、基部宽和圆形(球状)尖端。受影响的婴儿还经常在颈部区域有过多的皮肤(颈部皮肤)和颈部后部的低发际线(低后发际线)。
Noonan 综合征患者的面部特征往往会随着年龄的增长而以可预测的方式发生变化。在儿童后期,面部可能会显得比较粗糙,并开始呈现出更三角形的形状;此外,颈部变长,导致颈部蹼(翼状胬肉)显得更加明显和/或上背部和肩部(斜方肌)的大三角肌显得更加突出。在青春期,鼻梁变细变高,根部“捏”,基部宽,眼睛显得不那么突出。在成年期,特征性特征可能包括前额异常高的发际线;皱纹,异常透明的皮肤;鼻子和嘴唇之间有异常突出的褶皱(鼻唇沟)。此外,患有 Noonan 综合征的人在婴儿期可能有稀疏的头皮毛发,在儿童后期或青春期通常会变得更加毛茸茸或卷曲。许多受影响的人也有独特的眉毛,看起来高度拱形和/或“钻石形”。
许多患有努南综合征的新生儿出生体重正常。然而,在一些新生儿中,由于皮下组织层之间的液体异常积聚(皮下水肿),出生体重可能会增加。例如,手背和脚背肿胀(外周淋巴水肿)在 Noonan 综合征的新生儿中很常见;在这种情况下,影响手指的水肿可能导致指尖上的螺纹数量增加(异常皮纹)。这种水肿可能是由于某些淋巴管发育不当或发育迟缓(先天性淋巴发育不良)。
一些患有 Noonan 综合征的婴儿可能会遇到喂养问题,并且无法以预期的速度生长和增加体重(无法茁壮成长)。此外,患有这种疾病的儿童往往比他们的年龄矮,大约 20% 的儿童出现骨骼成熟延迟。大多数受影响的儿童在青春期前的生长速度(速度)相对正常;然而,在一些青少年中,通常在青春期经历的生长突增可能会减少或消失。患有 Noonan 综合征的男性成年平均身高约为 5 英尺 4 英寸(162.5 厘米),而患有该疾病的女性则约为 5 英尺(152.7 厘米)。患有这种疾病的人通常在生命的第二个十年结束时达到成年身高。生长模式受 NS 的分子遗传原因的影响。携带 RAF1 和 SHOC2 突变的 NS 人比其他基因型更矮,而携带 SOS1 和 BRAF 突变的人有更多的保留生长。
一些患有 Noonan 综合征的男性和女性也可能出现第二性征发育异常。在大约 60% 到 75% 的 Noonan 综合征男性中,一个或两个睾丸在出生前或出生后第一年未能降入阴囊(单侧或双侧隐睾)。如果不进行手术矫正,男性生殖细胞(精子)可能无法在睾丸内正常发育(精子发生不足),一些受影响的男性可能会出现不育症(不育)。其他患有 Noonan 综合征的男性可能会延迟但正常地获得第二性征(例如,睾丸、阴囊和阴茎的生长增加;面部和阴毛的出现等)。根据医学文献,在这种情况下,青春期可能平均延迟两年。其他患有 Noonan 综合征的男性可能会经历正常的青春期发育。即使没有隐睾病史,成年男性的生育能力似乎也有所下降。在患有这种疾病的女性中,获得第二性征(例如,阴毛的出现、乳房发育和月经)可能会轻微延迟,但通常是正常的。大多数患有努南综合征的女性生育能力正常。
许多Noonan综合征患者也有骨骼异常。大约 70% 的受影响儿童有明显的胸部畸形,其特征是胸骨(胸骨)上(上)部分异常突出和/或胸骨下(下)部分(鸡胸和/或胸骨)异常凹陷挖掘机,分别)。此外,胸部可能异常宽阔,乳头可能显得低矮。一些受影响的个体可能有额外的骨骼畸形,包括圆肩;肘部向外偏移(肘外翻);异常短的手指(短指),指尖钝;和/或脊柱前后弯曲和/或侧向弯曲(分别为脊柱后凸和/或脊柱侧凸)。当通过 DEXA 扫描评估时,患有 NS 的儿童的全身骨矿物质密度显着降低,这使他们面临骨折的风险。严重颈椎疾病的发生率也有所增加,包括颈椎管狭窄、Arnold-Chiari 畸形和脊髓空洞症。
大约三分之二患有 Noonan 综合征的婴儿在出生时也有心脏异常(先天性心脏缺陷)。在大约一半的此类病例中,受影响的婴儿会阻塞从心脏右下腔(心室)到肺部的正常血液流动(肺动脉狭窄)。在肺动脉狭窄患者中,心脏必须更加努力地将血液输送到肺部进行氧合。肺动脉狭窄导致的症状会有所不同,具体取决于狭窄的严重程度和任何其他相关发现。在一些严重的情况下,受影响的婴儿的心脏可能会在出生后立即开始扩大(即,在新生儿开始呼吸时)。在这种情况下,心脏可能无法有效地将血液(心力衰竭)泵送到肺部和全身。相关症状和发现可能包括由于异常低水平的循环氧(缺氧)、呼吸困难、腹部肿胀、喂养困难和/或其他异常导致的皮肤和黏膜呈蓝色变色(紫绀)。如果没有适当的治疗,可能会导致潜在的危及生命的并发症。在不太严重的肺动脉狭窄病例中,症状可能要到儿童后期才会出现。此类症状可能包括呼吸困难、易疲劳和/或其他异常。在其他情况下,肺动脉狭窄可能是轻微的并且可能不会出现症状(无症状)。
在大约 30% 的 Noonan 综合征婴儿中,分隔心脏两个上腔室(心房)的纤维分隔(隔膜)可能存在异常开口(房间隔缺损)。另有 20% 的先天性心脏缺陷患者可能存在分隔左右心室(室间隔)的隔板增大(肥大),并且在某些患者中,左心室壁(肥厚型心肌病)可能会增大(肥大)。不太常见的是,可能存在其他先天性心脏缺陷(例如,室间隔缺损、动脉导管未闭、房室管缺损)。根据医学文献,大多数 Noonan 综合征患者只有一个心脏缺陷。然而,例如,一些受影响的个体可能患有肺动脉狭窄并伴有房间隔缺损或肥厚性心肌病。
在患有先天性心脏缺陷的 Noonan 综合征患者中,约有 30% 发生房间隔缺损。在正常心脏中,出生时两个心房(卵圆孔)之间存在一个小开口。出生后不久,房间隔逐渐关闭并覆盖这个开口。然而,在患有房间隔缺损的婴儿中,房间隔可能无法正常关闭或可能在胎儿发育过程中畸形。结果,心房之间的开口在应该关闭后仍然存在很长时间,导致心脏右侧的工作量增加,以及右心室、右心房和主肺动脉的相关扩大。房间隔缺损的大小、位置和性质以及任何相关异常决定了症状的严重程度。
许多患有房间隔缺损的儿童没有任何症状。然而,在某些情况下,相关症状可能包括体重增加不佳、轻度生长迟缓,以及对反复呼吸道感染(例如肺炎)和心脏内膜(心内膜炎)和心脏瓣膜的细菌感染的易感性增加。在极少数情况下,严重受影响的儿童还可能出现呼吸困难、运动易疲劳、心力衰竭和/或心律不齐(心律失常)。
大约 20% 的患有心脏缺陷的受影响婴儿会出现肥厚型心肌病。在大多数情况下,这种异常扩大(肥大)会影响分隔左右心室的纤维分区的局部区域(前室间隔肥大);在其他情况下,整个隔膜和左心室壁可能会受到影响。肥厚型心肌病可能导致心输出量减少。相关症状和发现可能包括疲劳、劳累或运动期间短暂的昏厥发作(晕厥)和心力衰竭。如果没有适当的治疗,在某些情况下可能会导致危及生命的并发症。与其他患有 HCM 的儿童相比,患有 HCM 的 NS 患者在就诊时的风险状况更差,导致显着的早期死亡率(1 年时为 22%)。在极少数情况下,肥厚型心肌病也会在以后的生活中发展。
一些患有 Noonan 综合征的婴儿也可能有某些血管畸形,例如存在异常通道(瘘管),涉及向心肌供血的动脉(冠状动脉)。冠状动脉的轮廓也可以是扩张的(扩张的)和/或弯曲的(曲折的)。此外,一些受影响的婴儿可能有某些淋巴管畸形(先天性淋巴发育不良)。淋巴是一种含有白细胞(淋巴细胞)、脂肪和蛋白质的体液,在组织细胞之间的空间中积聚在血管外,并通过淋巴管流回血液中。在一些患有 Noonan 综合征的婴儿中,淋巴系统畸形可能包括淋巴组织内某些通道发育不全(发育不全),淋巴通过这些通道进入淋巴管;肺内淋巴管异常扩大(扩张)(肺淋巴管扩张);和/或扩大(扩张)肠淋巴管(肠淋巴管扩张),尤其是运输乳糜的血管,乳糜是在消化过程中从食物中吸收的乳状液体。肠道淋巴管扩张可能导致肠道吸收过程中蛋白质的损失(蛋白质丢失性肠病),某些循环白细胞水平异常低(淋巴细胞减少),以及含有过多脂肪的松散、恶臭的粪便(脂肪泻)。在青少年时期,一些 Noonan 综合征患者会出现下肢肿胀(淋巴水肿)。
在子宫内,一些受影响的婴儿可能在颈部皮肤下出现异常的囊性肿胀(囊性水肿)。此外,婴儿周围的羊水可能比平时多(羊水过多)。由于淋巴系统畸形和正常淋巴流入血流的相关阻塞,受影响的婴儿可能在某些组织中出现淋巴液异常积聚(淋巴水肿)。在某些情况下,水肿可能会影响全身的组织和腔体(胎儿水肿)。
大约 20% 到 33% 的 Noonan 综合征患者还存在各种凝血缺陷(凝血因子缺乏)、血液中循环血小板水平低(血小板减少症)和/或血小板功能不正常。血小板是一种特殊的血细胞,有助于预防和止血。受影响的个体的血液中某些物质(凝血因子)的含量可能较低,而这些物质对正常的凝血过程至关重要,这是一个复杂的过程,是止血(止血)所必需的。在 Noonan 综合征患者中,这种缺乏可能包括凝血因子 XI 和/或在某些情况下凝血因子 XII 和/或 VIII 水平低。一些受影响的个体可能患有血管性血友病;一种以凝血因子Ⅷ缺乏、出血时间延长和血小板粘附受损为特征的遗传性疾病。此外,在极少数情况下,受影响个体的尿液可能有异常的“鱼腥味”(三甲基氨基尿),这可能与血小板功能障碍有关。由于凝血因子缺乏、血小板功能障碍和/或血小板减少症,受影响的个体可能有异常且容易瘀伤和出血的病史。他们应该避免服用含阿司匹林的药物。
一些患有 Noonan 综合征的人也可能出现异常的皮肤变色。在大约四分之一的受影响个体中,可能存在色素痣(痣)。在极少数情况下,皮肤上可能会出现淡褐色或浅褐色斑块(咖啡牛奶斑)和/或黑色、深褐色或棕色“雀斑样”斑块(雀斑)。
多达 35% 的Noonan综合征患者也可能有轻度智力障碍。然而,许多受影响的人有正常的智商。 (智商)。此外,受影响的个体在获得需要协调精神和肌肉活动的技能(精神运动迟缓)、学习障碍和语言延迟方面可能会出现异常延迟,这可能是由于肌张力减退、说话困难和/或在某些个体中造成的,轻度听力损失。还报告了注意力不集中和执行功能的挑战。信息处理速度降低和其他认知领域功能相对完整是许多 NS 成人认知特征的特征。
三、病因
Noonan 综合征通常是由几种不同基因的异常(突变)引起的常染色体显性遗传疾病,主要有:PTPN11、KRAS、SOS1 RIT1 和 RAF1。在大约 50% 的受影响个体中发现了 PTPN11 突变;在不到 5% 的受影响者中发现了 KRAS 突变;在大约 13% 的 Noonan 综合征患者中发现了 SOS1 突变;在大约 5% 的 Noonan 综合征患者中观察到 RIT1 突变,在 5% 的患者中观察到 RAF1 突变。在少数病例中发现了与 Noonan 综合征相关的其他基因:NRAS、BRAF、MEK2、RRAS、RASA2、A2ML1 和 SOS2。新描述了与 SHOC2 和 CBL 突变相关的两种重叠情况。由 LZTR1 致病变异引起的 Noonan 综合征可以以常染色体显性或常染色体隐性方式遗传。
当只需要一个异常基因的一个拷贝来引起特定疾病时,就会发生显性遗传疾病。异常基因可以遗传自父母中的任何一方,也可以是受影响个体中新突变(基因改变)的结果。大约 50% 的受影响个体有受影响的父母。每次怀孕将异常基因从受影响的父母传给后代的风险为 50%。男性和女性的风险相同。
当个体从每个父母那里继承相同性状的相同异常基因时,就会发生隐性遗传疾病。如果一个人接受了一种正常基因和一种疾病基因,则该人将成为该疾病的携带者,但通常不会出现症状。两个携带者父母都通过缺陷基因并因此生育受影响的孩子的风险是每次怀孕的 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。一个孩子从父母双方那里获得正常基因并且在该特定特征上基因正常的机会是 25%。男性和女性的风险相同。
四、流行病学
Noonan 综合征似乎影响的男性多于女性,并且被认为影响大约 1,000 到 2,500 人中的 1 人。 然而,其他报告表明,这种疾病可能会影响普通人群中超过千分之一的新生儿。 自从 Noonan 综合征最初于 1883 年被报道(O. Kobylinski)并在 1963 年更详尽地描述(J.A. Noonan 和 D.A. Ehmke)以来,医学文献中讨论了 500 多名患者。 由于 Noonan 综合征的变异性极大,因此可能被低估或误诊,因此可能难以确定该疾病在普通人群中的真实频率。
五、鉴别诊断
以下疾病的症状可能与 Noonan 综合征的症状相似。比较可能有助于鉴别诊断:
面部皮肤 (Cardiofaciocutaneous,CFC) 综合征是一种极为罕见的遗传疾病,其特征是与 Noonan 综合征儿童相似的独特面部外观。其他主要特征可能包括异常稀疏、易碎、卷曲的头发;皮肤异常;出生时存在的心脏畸形(先天性心脏缺陷);生长延迟;和中度至重度智力障碍。心面皮肤综合征患者通常头部异常大(大头畸形)、前额突出、前额两侧异常变窄(双颞部收缩);短而上翘的鼻子,鼻根凹陷;眼部表现包括眼睑向下倾斜(睑裂)、眼睛间距过大(眼距过远)和/或上眼睑下垂(上睑下垂)。在大多数患者中,还存在先天性心脏缺陷,特别是从心脏右下腔(心室)到肺部的正常血流受阻(肺动脉瓣狭窄)和/或纤维分隔(隔膜)的异常开口) 分隔心脏的两个上腔室(心房)(房间隔缺损)。也可以发现心肌增厚(肥厚型心肌病)。此外,大多数患有这种疾病的人会出现生长迟缓、中度至重度智力障碍,以及在获得需要协调精神和肌肉活动的技能方面的异常延迟(精神运动迟缓)。面部皮肤综合征是由几个基因的突变引起的:BRAF、MEK1 和 2,以及 KRAS。 (有关更多信息,请选择“CFC 综合征”作为您在罕见病数据库中的搜索词。)
特纳综合征是一种染色体疾病,与努南综合征有一些相似之处。事实上,由于某些 Noonan 综合征患者表面上可能与 Turner 综合征患者相似(由于某些发现可能与这两种疾病有关,例如身材矮小、蹼状颈等),因此过去曾提到 Noonan 综合征如“男性特纳综合征”、“女性假特纳综合征”或“具有正常染色体的特纳表型”。然而,这两种疾病之间有许多重要的区别。 Noonan 综合征影响男性和女性,并且存在正常的染色体构成(核型)。只有女性会受到特纳综合征的影响,其特征是影响 X 染色体的异常。
患有特纳综合征的女性可能有一个短的蹼状颈部,后发际线较低;身材矮小;上眼睑下垂(上睑下垂)和/或眼睛间距过大(眼距过远);乳头间距大、倒置和/或发育不全(发育不全);先天性心脏缺陷,尤其是缩窄;和/或肾脏异常。在几乎所有情况下,都存在不能产生雌性激素雌激素的未成熟(条纹)卵巢。因此,正常的第二性征,如阴毛的出现、乳房发育和月经(原发性闭经)在青春期无法发育。几乎所有受影响的女性都不育。虽然智力通常是正常的,但有些人可能会在视觉空间关系方面遇到困难(例如,左右迷失方向)。
Costello综合征是一种罕见的遗传性疾病,其特征是出生后(产后)生长迟缓,导致身材矮小;独特的面部外观;颈部、手掌、手指和脚底过度松弛的皮肤;口腔(口周)、鼻孔(鼻孔)和肛门周围出现良性(非癌性)生长(乳头状瘤);和轻度至中度智力障碍。患有这种疾病的新生儿可能在全身组织中出现淋巴液异常积聚(全身性淋巴水肿)和高出生体重。此外,受影响的婴儿通常有严重的喂养和吞咽困难,并且可能无法以预期的速度生长和增加体重(无法茁壮成长)。
与该疾病相关的特征性颅面异常可能包括异常大的头部(大头畸形)和宽前额;一个大而凹陷的鼻根;异常宽的鼻孔;可能覆盖眼睛内角的皮肤褶皱(内眦赘皮);耳朵位置低,耳垂大而厚;和/或异常厚的嘴唇。其他身体特征可能包括手掌和脚底出现干燥、硬化、增厚的皮肤(掌跖角化过度)和/或手掌和脚底出现异常深的折痕。许多受影响的个体也可能有与 Noonan 和 CFC 综合征相似的先天性心脏缺陷。更明显的是在大约三分之一的地方出现了不寻常和混乱的节奏。大多数 Costello 综合征病例是零星发生的,没有该疾病的家族史,是由 HRAS 突变引起的。 (有关这种疾病的更多信息,请在罕见病数据库中选择“Costello”作为您的搜索词。)
多发性巨细胞病变是涉及颌骨和软组织内的某些大细胞(巨细胞)的异常囊肿(病变)。它们存在于 Noonan 综合征和其他重叠疾病(如 CFC 综合征)中,通常在头 20 年内被诊断出来。
神经纤维瘤病-Noonan 综合征的特征是发生 I 型神经纤维瘤病并伴有 Noonan 综合征的某些表现。相关症状和发现可能包括神经和皮肤的多个良性肿瘤、身材矮小、颈蹼(翼状胬肉)、肌肉无力和/或学习障碍。受影响的个体还可能具有与 Noonan 综合征相关的某些颅面异常,包括上眼睑下垂(上睑下垂)、低位耳朵和/或鼻子和嘴唇之间异常突出的褶皱(鼻唇沟)。此外,可能存在常见于 Noonan 综合征的先天性心脏缺陷,例如心脏右下腔(心室)的正常血液流出受阻(肺动脉狭窄)和/或纤维分隔异常开口。隔膜)在心脏的上腔室(心房)之间(房间隔缺损)。神经纤维瘤病-努南综合征可能是由于两种疾病在同一个体中的偶然发生,可能是 I 型神经纤维瘤病的表型,或者可能是由 NF1 基因突变引起的单独疾病实体,但没有某些特征性特征NF1。
Noonan伴多发性痣综合征(NSML,以前称为 LEOPARD 综合征)是一种罕见的遗传性疾病,其特征是皮肤、心脏结构和功能、内耳、头部和面部(颅面)区域和/或生殖器。在患有这种疾病的个体中,症状和身体特征的范围和严重程度可能因人而异。
一些最常见的特征包括雀斑(皮肤上有多个黑色或深棕色斑点);心电图传导缺陷(电活动异常和心脏适当收缩的协调);肥厚型心肌病,眼距过远(眼距宽);肺动脉狭窄(心脏右心室血液正常流出受阻);生殖器异常(男孩通常睾丸未降(隐睾);生长缓慢导致身材矮小、发育迟缓;以及由于内耳功能障碍导致的耳聋或听力丧失(感觉神经性耳聋)。NSML 是一种常染色体显性遗传疾病,由以下原因引起两个基因之一的突变:PTPN11 或 RAF1。
六、诊断
在某些情况下,根据胎儿超声检查的结果,可能会在出生前(产前)怀疑 Noonan 综合征,这是一种专门的成像技术,其中声波用于创建发育中胎儿的图像。 Noonan 综合征的诊断可能是由于母体血清三重筛查异常,检测到羊膜囊内胎儿周围的羊水过多(羊水过多),颈部区域存在由扩张的淋巴管组成的异常囊性肿胀(囊性hygroma)、结构性心脏差异、其他胎儿异常以及正常染色体构成(核型)的确认。然而,在许多情况下,Noonan 综合征是在出生或婴儿早期诊断的,这是基于全面的临床评估、特征性身体检查结果的识别和各种专门测试。如果产前怀疑 Noonan 综合征,可通过羊水或无细胞胎儿 DNA 分析进行分子遗传学检测。
需要注意的是,在某些情况下,仅具有与 Noonan 综合征相关的轻微、微妙特征的个体可能不会得到诊断。专门诊断和治疗心脏异常的医生(心脏病专家)应该怀疑任何患有先天性肺动脉瓣狭窄的人可能患有 Noonan 综合征。因为在这种情况下可能难以确认 Noonan 综合征(特别是如果没有该疾病的家族史),因此强烈认为 Noonan 综合征是任何患有肺动脉瓣狭窄和某些眼部异常的个体的可能诊断,即使在较轻的病例(例如,上睑下垂、内眦赘皮、眼距过远)。此外,在这种情况下,应检查所有直系(一级)亲属是否存在可能与 Noonan 综合征相关的轻度面部异常和心脏缺陷。
在许多患有该疾病的个体中,某些先进的成像技术和实验室测试可用于检测、确认和/或表征可能与 Noonan 综合征相关的特定异常。
与 Noonan 综合征相关的先天性心脏缺陷可以通过彻底的临床检查和允许医生评估心脏结构和功能的专门测试来检测和/或确认。临床检查可能包括医生使用听诊器评估心音和肺音。在肺动脉狭窄的轻度无症状病例中,最初可以通过在这种听诊器评估期间听到的异常心脏杂音来检测这种情况。
专门的心脏检查可能包括心电图 (EKG)、超声心动图和/或心导管检查。记录心肌电活动的心电图可能会显示异常的电模式(例如,电轴左偏、左前半传导阻滞、深 S 波)。在超声心动图中,声波指向心脏,使医生能够研究心脏功能和运动。在心导管插入术中,将一根小的空心管(导管)插入大静脉并穿过通向心脏的血管。
该程序允许医生确定通过心脏的血流速度、测量心脏内的压力和/或彻底识别解剖异常。此外,医生还可能密切评估呼吸(通气)能力,因为相关的心脏缺陷可能导致肺部血液供应不足和呼吸困难。
可以进行专门的血液检查以检测潜在的凝血因子缺乏和/或血小板功能障碍。
相关基因突变的分子遗传学检测可用于确诊和产前诊断。
七、治疗
Noonan 综合征的治疗针对每个人明显的特定并发症。治疗可能需要专家团队的协调努力。儿科医生、诊断和治疗心脏异常的医生(心脏病专家)、诊断和治疗血液和造血组织疾病的医生(血液科医生)、诊断和治疗生长障碍的医生(内分泌科医生)和/或其他医疗保健专业人员可能需要系统和全面地规划受影响儿童的治疗。
在一些患有先天性心脏缺陷的个体中,可能需要使用某些药物、手术干预和/或其他技术进行治疗。在这种情况下,执行的任何外科手术将取决于解剖异常及其相关症状的位置、严重程度和/或组合。在决定外科手术时,必须考虑可能存在的心脏、动静脉和/或淋巴管畸形。例如,在对淋巴管瘤进行某些类型的手术期间,乳糜可能从体内最大的淋巴通道(胸导管)逃逸到颈部和横膈膜之间的腔(胸腔)的风险增加,可能导致生命- 威胁性并发症(乳糜胸)。
对于同时患有血小板减少症、血小板功能障碍和/或凝血因子缺乏症的患者,医生、牙医和/或其他医护人员可能会在手术前推荐某些预防措施或在手术期间采取某些支持措施,以预防、降低风险或控制异常出血。
呼吸道感染应及时积极治疗。由于心脏内膜(心内膜炎)和心脏瓣膜的细菌感染风险可能增加,因此患有某些心脏缺陷的受影响个体可能会在任何外科手术(包括牙科手术,例如拔牙)之前接受药物治疗。
对于患有隐睾症的受累男性,应进行手术将未降的睾丸移入阴囊并将其固定在固定位置(睾丸固定术)。此类手术通常在 12 至 24 个月大时进行,以帮助预防相关不孕症的风险。
此外,适当的支持措施可用于受影响的淋巴水肿个体。
早期干预可能对帮助 Noonan 综合征儿童发挥潜能很重要。可能对受影响儿童有益的特殊服务可能包括特殊补救教育、言语治疗、物理治疗和其他医疗、社会和/或职业服务。 Noonan 综合征患者的身材矮小可以用生长激素治疗,生长激素已被证明可以提高最终成年身高。
建议对受影响的个人及其家人进行遗传咨询。如前所述,对确诊患者的家庭成员进行彻底的临床评估可能很重要,以检测可能与 Noonan 综合征相关的任何症状和身体特征。该疾病的其他治疗是对症治疗和支持治疗。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Allanson JE. Noonan Syndrome. In: The NORD Guide to Rare Disorders, Philadelphia, PA: Lippincott, Williams and Wilkins; 2003: 722-723.
Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, Blair E, Borck G, Brinkmann J, Craigen W, Dung VC, Emrick L, Everman DB, van Gassen KL, Gulsuner S, Harr MH, Jain M, Kuechler A, Leppig KA, McDonald-McGinn DM, Can NTB, Peleg A, Roeder ER, Rogers RC, Sagi-Dain L, Sapp JC, Schäffer AA, Schanze D, Stewart H, Taylor JC, Verbeek NE, Walkiewicz MA, Zackai EH, Zweier C, Zenker M, Lee B, Biesecker LG, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. 2018;20:1175–85. PMID: 29469822.
Prendiville TW, Gauvreau K, Tworog-Dube E, Patkin L, Kucherlapati RS, Roberts AE, Lacro RV. Cardiovascular disease in Noonan syndrome. Arch Dis Child. 2014 Jul;99(7):629-34. Epub 2014;Feb 17. PMID: 24534818
Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013 Jan 26;381(9863):333-42. doi: 10.1016/S0140-6736(12)61023-X. Epub 2013;Jan 10. Review.PMID:23312968
Briggs BJ, Dickerman JD. Bleeding disorders in Noonan syndrome. Pediatr Blood Cancer. 2012;58(2):167-72.
Malaquias AC, Brasil AS, Pereira AC, et al. Growth standards of patients with Noonan and Noonan-like syndromes with mutations in the RAS/MAPK pathway. Am J Med Genet. 2012;Nov;158A(11):2700-6.
Smpokou P, Tworog-Dube E, Kucherlapati RS, Roberts AE. Medical complications, clinical findings, and educational outcomes in adults with Noonan syndrome. Am J Med Genet A. 2012 Dec;158A(12):3106-11. doi: 10.1002/ajmg.a.35639. Epub 2012 Nov 19. PMID: 23165751
Wilkinson JD, Lowe AM, Salbert BA, et al. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: A study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2012;164(3):442-8.
Wingbermühle E, Roelofs RL, van der Burgt I, et al. Cognitive functioning of adults with Noonan syndrome: a case-control study. Genes Brain Behav. 2012;Oct;11(7):785-93.
Baldassarre G, Mussa A, Dotta A, et al. Prenatal features of Noonan syndrome: prevalence and prognostic value. Prenat Diagn. 2011;31(10):949-54.
Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;Feb;25(1):161-79.
Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126(4):746-59.
Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, SeidenbergMS Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav. 2009;8(3):275-82.
Noordam C, Peer PGM, Francois I, De Schepper J, van der Burgt I, Otten BJ . Long-term GH treatment improves adult height in children with Noonan syndrome with and without mutations in protein tyrosine kinase phosphatase, non-receptor-type 11. Eur J Endocrinol. 2008;159: 203-6.
Osio D, Dahlgren J, Wikland KA, et al. Improved final height with long-term growth hormone treatment in Noonan syndrome. Acta Paediatr. 2005;94:1232-7.
Allanson JE, Roberts AE. Noonan Syndrome. 2001 Nov 15 [Updated 2019 Aug 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1124/ Accessed August 20, 2019.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#Noonan综合征##罕见病#
110
#流行病#
70
#综合征#
92