
2例鼻出血引发的思考
2023-02-03 魏佳等 苏州大学附属儿童医院 “检验医学”公众号 发表于陕西省

该病的发病机制主要涉及血小板膜糖蛋白Ⅱb/Ⅲ a(GP Ⅱ b/Ⅲ a)的结构异常和功能障碍,活化的血小板无法通过纤维蛋白原结合相互聚集。
前 言
血小板无力症是一种罕见的常染色体隐性遗传的出血疾病,临床主要表现为自幼发生皮肤黏膜出血或创伤性出血,出血表现存在异质性。该病的发病机制主要涉及血小板膜糖蛋白Ⅱb/Ⅲ a(GP Ⅱ b/Ⅲ a)的结构异常和功能障碍,活化的血小板无法通过纤维蛋白原结合相互聚集。
目前血小板聚集功能检查和血小板GP Ⅱ b/Ⅲ a复合物数目或质量异常最具诊断意义,基因诊断并非必备条件。由于对该病认识不够,导致初诊时间较起病时间往往延迟很多,并且多数医院缺乏诊断该疾病的实验室条件,因此血小板无力症在我国的早期诊断率较低,误诊率和病死率较高。
案例经过
案例1
患者1既往确诊“血小板功能障碍”9年余,长期反复出现鼻出血和皮肤黏膜瘀斑,在外院具体诊疗过程不详。本次该患者因鼻出血加重来我院就诊,由于出血量大转入ICU治疗。经与血液科和检验科沟通,进行一系列血小板功能实验筛查。结果发现该患者二磷酸腺苷(ADP)诱导的血小板最大聚集率为0%,瑞斯托霉素诱导的血小板最大聚集率为46%;同时血小板膜糖蛋白GP Ⅱb、GP Ⅲa表达缺失。经进一步分子遗传学检查发现该患者ITGA2B基因2处突变,明确了血小板无力症的诊断。次年该患者因月经来潮,行经不止,长期依赖血小板输注,最终选择进行造血干细胞移植治疗。
案例2
患者2既往因反复鼻出血6天余,出血量较多,不能自止,来我院耳鼻喉科进行手术治疗。住院期间复查多次凝血常规和凝血因子全套项目,结果均无异常,术后正常出院。经5月余,该患者又因皮肤出现瘀点瘀斑来我院血液科门诊就诊。经临床与检验科沟通,检验科结合其皮肤易出现青紫瘀斑的既往史,建议再加做血小板聚集试验的筛查。结果发现该患者ADP与花生四烯酸(Ara)诱导的血小板最大聚集率均<1%,瑞斯托霉素诱导的血小板最大聚集率为17.7%。后经分子遗传学检查发现该患者ITGB3基因有一处变异。但因家长拒绝入院治疗,后续治疗过程不详。
临床案例分析
案例1
患儿女,11岁7个月,江苏宿迁人,因“鼻出血3天伴刺激性呛咳”于2020年11月14日入院。患儿3天前患儿无明显诱因出现鼻出血,血液呈鲜红色,出血较多,至当地耳鼻喉科住院治疗,仍出血不止,出血量大引起身体抽搐、小便失禁和面色苍白,遂来我院急诊就诊,以“血小板功能障碍”收入我院ICU治疗。
患儿有血小板功能障碍、鼻出血和皮肤黏膜瘀斑等既往史。血小板聚集试验结果显示血小板聚集能力低下。同时,GP Ⅱb、GP Ⅲa表达缺失。基因检测发现,ITGA2B c.471dupC(p.Val58fs)插入移码突变及c.1750C>T(p.Arg584Ter)无义突变。结合其临床出血表现、实验室检查和基因检测结果,确诊为血小板无力症。
案例2
患儿,男,4岁7个月,江苏苏州人,因“反复鼻出血及皮肤瘀斑瘀点半年”于2022年5月19日至我院血液科门诊就诊。患者半年前无明显诱因下出现鼻出血,出血量多,不能自止,伴随皮肤黏膜出现瘀点瘀斑,久不消退,期间多次至当地医院及我院耳鼻喉科就诊,疗效欠佳,来我院血液科门诊就诊,门诊拟“紫癜”进行检查治疗。
血小板聚集试验结果显示血小板聚集能力低下。基因检测发现,ITGB3一处来自父母双方的移码突变。结合其临床出血表现、实验室检查和基因检测结果,诊断为血小板无力症。
检验案例分析
案例1
2020年11月14日入院
2020年11月15日晨常规行空腹检测:血常规:WBC 13.16(*10^9/L)、Hb 57g/L、PLT 216(*10^9/L)、N% 75% CRP 13.52mg/L。血气分析及电解质:PH 7.47、二氧化碳分压28.8mmHg、氧分压73.6 mmHg、碳酸氢根20.6mmol/L、剩余碱-3.1mmol/L、钠138mmol/L、钾3.6mmol/L、氯105mmol/L、钙1.01mmol/L、乳酸5.1mmol/L。
入院后患儿完善其他实验室检查:
凝血常规:PT 13.5s、INR 1.19、APTT 26.5s、Fib 2.23g/L、TT 18.4s、ATⅢ 81.5%、DDi 970ug/L(↑)。
血栓弹力图:凝血因子活性(R):3.8min、纤维蛋白原水平(Angle):43.4deg、血凝块强度(G):924d/sc、血小板聚集功能(MA):15.6mm。
生化全套:CRP 15.72mg/L、ALT 6.5U/L、ALB 34.2g/L、肌酐43.1umol/L、免疫球蛋白0.41g/L、前白蛋白147mg/L。
血小板聚集试验:ADP诱导的血小板最大聚集率:0(正常值59.1-98.3%),瑞斯托霉素诱导的血小板最大聚集率:46%(正常值58%-76%)。
流式细胞免疫荧光分析:血小板膜糖蛋白检测结果为GP Ⅱb、GP Ⅲa表达缺失。
全外显子组高通量测序检测技术:ITGA2B c.471dupC(p.Val58fs)插入移码突变(来自母亲)及c.1750C>T(p.Arg584Ter)无义突变(来自父亲)。
案例2
2022年5月19日血液科门诊常规检查:
血常规:WBC 8.07(*10^9/L)、RBC 4.04(*10^12/L)、Hb 112g/L、PLT 206(*10^9/L)、N% 43.4%、L% 43.9%。
2022年5月25日血液科门诊完善凝血相关项目的检查。
凝血常规:PT 11.2s、INR 1.19、APTT 23.8s、Fib 3.716g/L(↑)、TT 17.1s、AT3 113.7%、DDi 1500ug/L(↑)。
凝血因子全套:因子Ⅱ121.2%(↑);因子Ⅴ125.6%(↑);因子Ⅶ95%;因子Ⅷ205.1%(↑); Ⅸ因子90.2%; Ⅹ因子100.5%; Ⅺ因子123.6%; Ⅻ 77.9%; 抗Ⅹa(低分子肝素) 0;抗Ⅹa(肝素) 0。
血栓四项:TM 9.5IU/mL;t-PAI-C 3.2ng/mL;TAT 14.5ng/mL(↑);PIC 5.83ng/mL(↑)。血小板聚集试验:ADP和Ara诱导的血小板最大聚集率均<1%(ADP正常值:59.1-98.3%;Ara正常值:63.2-98.3%;见图1-2),瑞斯托霉素诱导的血小板最大聚集率为17.7%(正常值58-76%;见图3)。
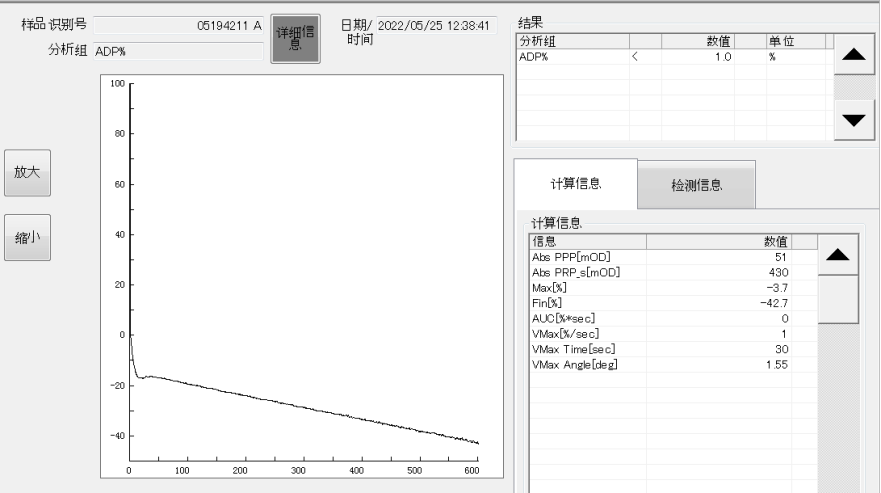
图1 血小板聚集试验中ADP诱导的血小板聚集结果

图2 血小板聚集试验中Ara诱导的血小板聚集结果
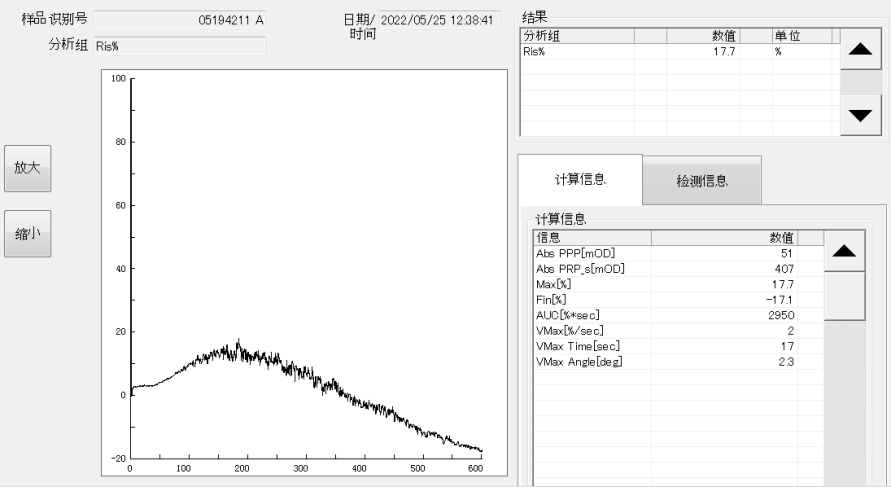
图3 血小板聚集试验中Ris诱导的血小板聚集结果
2022年5月26日根据血小板聚集功能试验的结果完善基因筛查项目:
采用全外显子组高通量测序检测技术,该患者样本检测到基因ITGB3的一处来自父母双方的移码突变(图4)。ITGB3:NM_000212.2:exon4:c.401del:p.E134Gfs*10变异:致病(PVS1+PM3+PM2_Supporting)
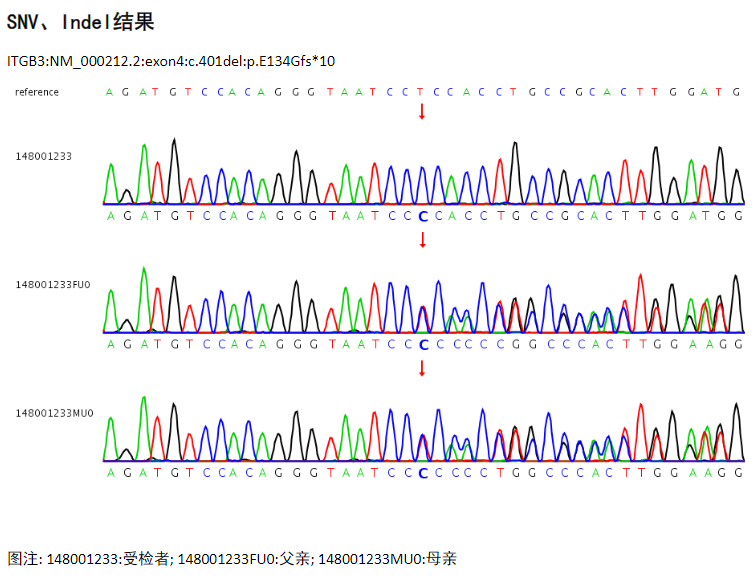
图4 全外显子组高通量测序检测结果(家系)
知识拓展
血小板无力症(Glanzmann thrombasthenia, GT)是一种罕见的遗传性血小板功能缺陷性疾病,为常染色体隐形遗传病。这是一种由血小板整合素α2b亚单位(integrin subunit α2b, ITGA2B)或整合素β3亚单位(integrin subunit α2b, ITGB3)基因缺陷引起膜糖蛋白Ⅱb/Ⅲa复合物(glycoprotein Ⅱb/Ⅲa, GP Ⅱb/Ⅲa)结构或数量异常引起血小板聚集功能缺陷或血凝块收缩减弱。
临床上主要表现为自发性出血或创伤后出血不止,出血表现存在异质性。患者常表现为皮肤瘀点瘀斑(>70%)、鼻出血(约69.04%)、牙龈出血(约36.08%)、外伤后出血(约19.59%)及90.57%初潮后女性患者可表现为月经过多。
GT初筛实验主要包括血涂片、出血时间(bleeding time, BT)和血块收缩实验等,进一步评估包括血小板功能实验检测或分子遗传学检查。血小板功能实验包括血小板聚集试验、流失细胞数/蛋白质印迹检测GP Ⅱb(CD41)、GP Ⅲa(CD61)表达水平。
患者通常表现为二磷酸腺苷(adenosine diphosphate, ADP)、花生四烯酸(arachidonic acid, Ara)等多种生理诱导剂聚集率减低或缺失,瑞斯托霉素(Ristocetin, Ris)诱导的聚集率正常。
通过上述实验室检查仍未确诊的病例,国际血栓和止血学会(International Society of Thrombosis and Haemostasis, ISTH)指南建议进行分子遗传学检测,重点筛选ITGA2B、ITGB3基因等。
目前GT数据库(http://sinaicentral.mssm.edu/intranet/research/glanzmann/search)登记的ITGA2B基因突变有256种,ITGB3基因突变有164种。致病的基因突变可发生在GP Ⅱb的β螺旋、Calf1区和Calf2区,GP Ⅲa的PSI区、βⅠ区、EGF(1-4)区和βTD等,其中GP Ⅱb β螺旋区和GP Ⅲa βⅠ区的突变最为常见。不同结构域突变对GP Ⅱb/Ⅲa的生物合成、转运及功能等影响并不相同,这也是导致临床表现各异的主要原因。
GT的出血程度与血小板表面GP Ⅱb/Ⅲa的缺陷程度无明显相关性,有的患者即使在血小板表面检测不到GP Ⅱb/Ⅲa,也仅有轻微出血,而有的患者虽然血小板表面GP Ⅱb/Ⅲa无明显减少,却存在严重出血倾向。这与不同患者之间个体情况、机体其他止血因素代偿状况有关,但也与细胞的自我代偿及自我修复有关。
因此,GT患者应尽可能避免外伤和手术。目前针对GT尚没有有效的治疗指南,主要的治疗手段主要包括输注血小板外、选择性应用rFⅦa及女性月经期及孕期应用雌激素治疗。对于严重出血、依赖血小板输注的GT患者进行HLA相合的同胞或无关供者异基因骨髓移植具有潜在的治疗价值。
虽然造血干细胞移植本身风险大,但总体的预后较好。也可通过首先确GP Ⅱb/Ⅲa基因缺陷再选择性进行基因修复是目前公认的可能根治GT的方法,也是目前国际上GP Ⅱb/Ⅲa复合物研究的热点领域之一。
此外,遗传学血小板无力症也可选择产前早期诊断。血小板膜GP在胎儿期即完全生成,在孕18-26周时脐血血小板膜GP Ⅱb/Ⅲa的表达已与成人相当。但该种产前基因诊断对胎儿有一定的损伤危险性,应在有一定条件和经验的医疗单位中进行。
我国对GT的研究和治疗水平正在逐步提升阶段,但对于这类罕见疾病的早期诊断和治疗仍面临着许多挑战。由于不同地区的医疗资源和诊疗水平存在明显差异,同时该病的罕见性令研究人员很难开展大规模的临床试验,因此国际上推荐建立GT登记中心、创建可公开访问的数据库等措施来保证GT患者及时获得高质量的医疗服务和改善预后。
案例总结
血小板无力症是一类罕见的血小板功能障碍常染色体隐性遗传性疾病,此类疾病均具有终生出血的倾向,通常在儿童期即出现临床表现。由于对该病认识不够,导致初诊时间较起病时间往往延迟很多,目前国内报道诊断最早的为出生3个月后,多数患儿均在一岁以后确诊。
本案例中患儿1和患儿2均有鼻出血严重就诊病病史,同时伴有皮肤反复出现瘀点瘀斑。尤其女性患儿1进入青春期后,出现了典型的月经过多表现。
本案例中两名患儿血常规中血小板计数和血小板分布宽度均正常。目前血小板聚集功能检查和血小板GP Ⅱb/Ⅲa复合物数目或质量异常最具诊断意义。
本案例中两名患儿血小板聚集试验中ADP和Ara诱导血小板最大聚集率均<1%。然而Ris诱导的最大聚集率也有不同程度的减低,鉴于两名患者在进行血小板聚集试验前已接受止血和手术治疗,因此该结果可能受患者体内潜在的药物影响、营养状况等影响导致的。
根据血小板表面GP Ⅱb/Ⅲa的数量血小板无力症可分3型。Ⅰ型血小板表面GP Ⅱb/Ⅲa含量小于正常的5%活化的血小板不能结合FIB,血块缺乏回缩反应;Ⅱ型血小板表面GP Ⅱb/Ⅲa为正常的10%-20%活化的血小板可少量结合FIB,血块回缩异常;Ⅲ型为变异型(结构异常)血小板表面GP Ⅱb/Ⅲa为正常的50%-100%,但活化的血小板不能结合或仅少量结合FIB,血块回缩从缺乏到正常。
患者1检查了血小板膜糖蛋白GP Ⅱb/Ⅲa复合物数目发现表达缺失,属于Ⅰ型。此外,基因诊断并非是诊断血小板无力症的必备条件。从方法学的角度出发,PCR技术结合测序的方法对于一些大片段缺失和突变存在漏检现象。
还有蛋白翻译后修饰异常、空间结构异常、整合素亚单位的转运异常、整合素的信号传导系统的异常等都能导致血小板无力症的发生,却不一定能找到基因突变。因此,仅仅通过对外显子区测序分析或cDNA测序分析,一些遗传性血小板无力症的基因缺陷还是不能被完全发现。
本案例中两名患儿均是经典的ITGA2B、ITGB3基因发生突变,且都为杂合突变。
在检验工作中遇到血小板计数正常,但有出血倾向临床表现的病例,应积极与临床沟通,推荐血小板功能实验来筛查血小板功能障碍的一系列疾病。在得到血小板聚集低下的结果后,并进一步建议进行基因检测。总之,临床异常的出凝血表现,联合血小板功能试验、基因检测等实验室项目,可以对早期诊断和治疗罕见的遗传性血小板相关疾病有重大的临床意义。
专家点评
点评专家:苏州大学附属儿童医院 血液科 卢俊教授、检验科 邵雪君教授
该案例为本院的真实的临床案例,患者1为诊断多年血小板功能障碍,但长期反复出现鼻出血和皮肤瘀点瘀斑,在外院多次对症止血治疗确未明确病因。因出血量大,病情逐渐加重来我院就诊,经我院临床与微生物检验医师沟通,进行血小板聚集功能试验、血小板GP Ⅱb/Ⅲa表达量检测、血栓弹力图、基因检测等一系列检查,最终确诊了血小板无力症。因患者1青春期月经初潮后,行经不止,又在我院进行了造血干细胞移植手术,解决了患者长期进行血小板输注的痛苦问题。患者2先前由于严重鼻出血来我院耳鼻喉科住院手术治疗,后因出现皮肤瘀斑瘀点的症状来我院血液科门诊治疗。结合该病例前期鼻出血和皮肤易青紫的既往病史,我院检验医师与临床积极沟通,建议进行血小板聚集试验筛查。结果发现血小板聚集减弱后又进行了基因检测,最终快速确诊了血小板无力症。对于少见的凝血与血栓疾病的诊断,临床与检验的沟通、选择合适的凝血项目筛查至关重要。本案例真实、可靠,是检验与临床沟通,检验科为临床疑难病例的诊断提供良好服务的典型病例,值得分享。
参考文献
[1]甘芳宴,屈晨雪.血小板无力症发病机制研究进展[J].临床检验杂志,2019,37(9):686-690.
[2]黎海燕,李保才,李丽君.血小板无力症的诊治新进展[J].临床输血与检验,2022,24(2):244-249.
[3] Nurden AT, Pillois X. ITGA2B and ITGB3 gene mutations associated with Glanzmann thrombasthenia[J]. Platelets, 2018, 29(1):98-101.
[4] Botero JP, Lee K, Branchford BR, et al. Glanzmann thrombasthenia: genetic basis and clinical correlates[J]. Haematologica, 2020, 105(4):888-894.
[5]苗林子,甘芳宴,龚岩,等.八例血小板无力症的基因突变特征分析[J].中华医学杂志,2018,98(30):2418-2423.
[6] ZHOU L, JIANG M, SHEN H, et al. Clinical and molecular insights into Glanzmann's thrombasthenia in China[J]. Clinical Genetics: An International Journal of Genetics in Medicine, 2018, 94(2):213-220.
[7] Huang JS, Li X, Shi XF, et al. Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting[J]. J Hematol Oncol, 2019, 12(1):26.
[8] Poon MC. The Use of Recombinant Activated Factor VII in Patients with Glanzmann's Thrombasthenia[J]. Thromb Haemost, 2021, 121(3):332-340.
[9]李建琴,王兆钺,胡绍燕,等.遗传性血小板无力症的发病机制及产前诊断[J].临床儿科杂志,2016(2):132-135.
[10] Minno GD, Zotz RB, D'oiron R, et al. The international, prospective Glanzmann Thrombasthenia Registry: treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia[J]. Haematologica, 2015, 100(8):1031-1037.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







还需要努力开发安全但高效的抗克隆疗法,并等待目前评估淀粉样蛋白靶向疗法的研究结果。
56