
Cardiovasc Res 天津医科大学朱毅教授/上海交通大学付成来研究员发现抑制IP6K1缓解心肌缺血再灌注损伤的作用机制
2024-02-08 论道心血管 论道心血管 发表于陕西省

研究发现焦磷酸肌醇5-InsP7是脂联素的内源性抑制剂,抑制IP6K1 (5-InsP7合成关键酶)通过上调血浆脂联素来减轻心肌缺血再灌注损伤。
心血管事件是导致死亡的主要原因之一,急性心肌梗死后的缺血再灌注损伤是目前治疗的难点。脂联素主要由脂肪细胞合成并分泌入血,具有抗炎、抗凋亡、抗氧化应激等多种心血管保护作用。增加脂联素的血浆水平可以减轻缺血再灌注导致的心肌损伤。由于脂联素的血浆浓度高、代谢快等特点,很难通过外源补充脂联素来维持有效浓度和作用时间。因此,上调内源性脂联素的表达则是有效的治疗策略。
2024年1月22日,天津医科大学朱毅教授与上海交通大学医学院附属新华医院付成来研究员合作在Cardiovascular Research在线发表了题为“Depleting inositol pyrophosphate 5-InsP7 protected the heart against ischemia-reperfusion injury by elevating plasma adiponectin”的研究论文,发现焦磷酸肌醇5-InsP7是脂联素的内源性抑制剂,抑制IP6K1 (5-InsP7合成关键酶)通过上调血浆脂联素来减轻心肌缺血再灌注损伤,研究结果提示IP6K1可能是潜在的心血管疾病的干预靶点。

研究发现,抑制5-InsP7的表达降低心血管疾病风险因素。为了探索抑制5-InsP7的表达能否减轻心肌缺血再灌注损伤,研究团队利用WT和IP6K1 (5-InsP7合成关键酶)敲除小鼠构建心肌缺血再灌注损伤模型。实验结果显示,敲除IP6K1显著减轻心肌缺血再灌注损伤。研究人员还发现,敲除IP6K1上调血浆脂联素。
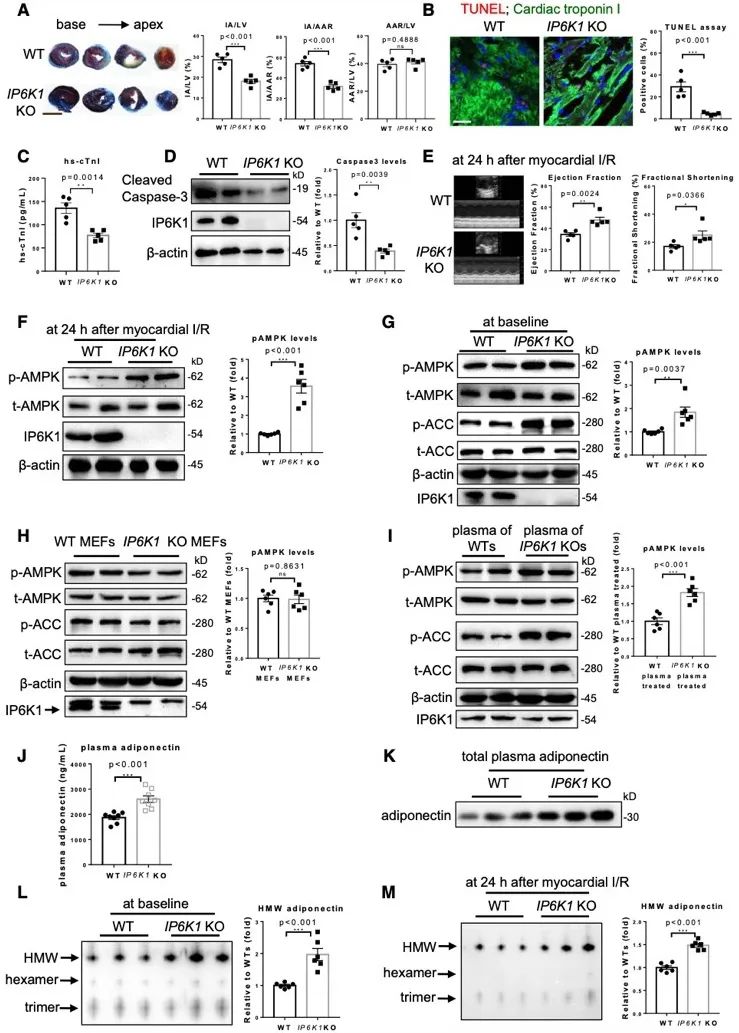
由于血浆脂联素主要来源于脂肪细胞,研究团队构建脂肪特异性IP6K1敲除小鼠,验证了敲除IP6K1通过上调脂联素来缓解心肌缺血再灌注损伤。
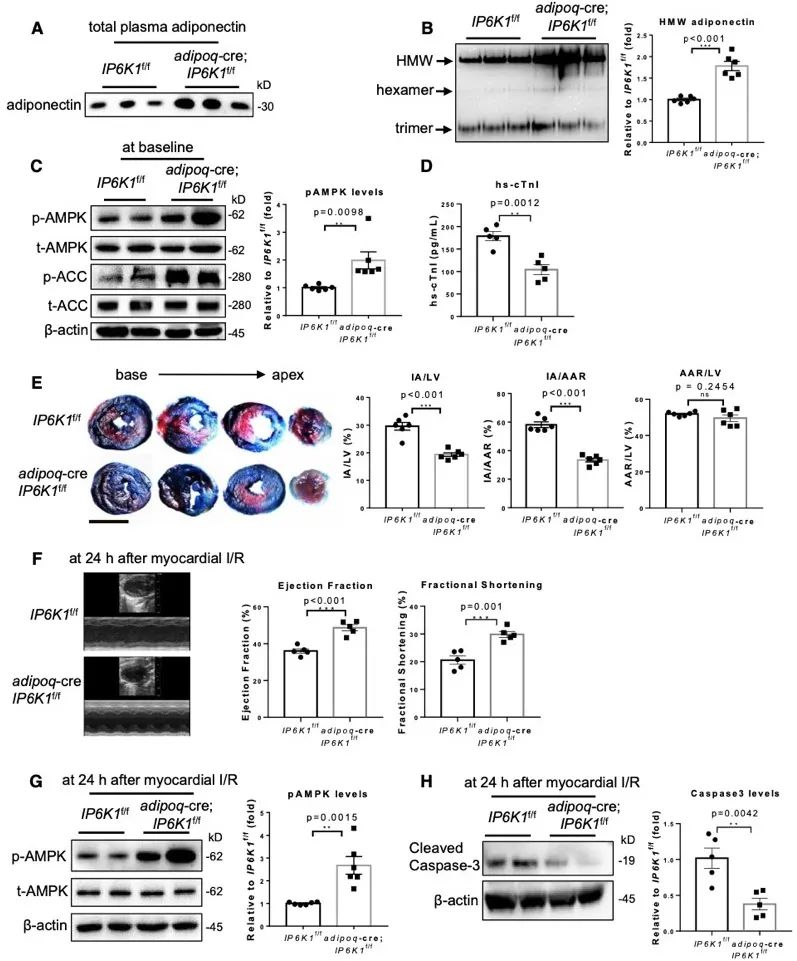
为了探究IP6K1和其产物5-InsP7调控脂联素的分子机制,作者利用免疫沉淀、蛋白质谱分析和western blot等方法发现并证实IP6K1结合脂联素。
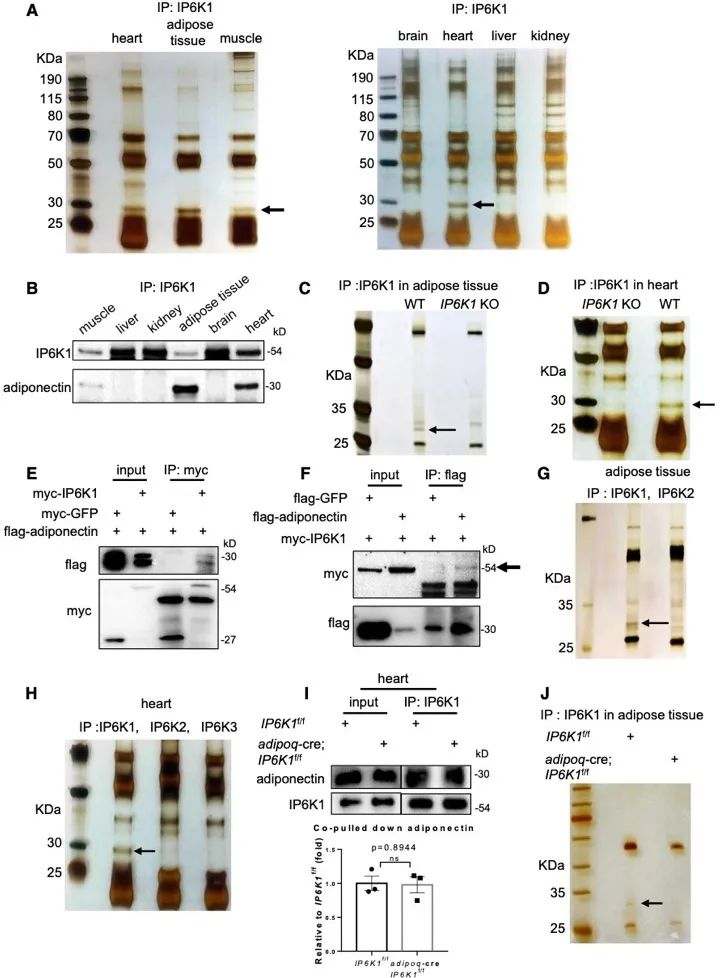
脂联素在脂肪细胞内的合成和分泌受内质网分子伴侣ERp44的调控。作者发现,IP6K1通过其产物5-InsP7促进了脂联素与内质网分子伴侣ERp44之间的相互作用。

DsbA-L和Ero1-Lα也是调控脂联素合成和分泌的关键蛋白。作者发现,IP6K1通过其产物5-InsP7促进DsbA-L与Ero1-Lα的结合。上述研究结果提示,IP6K1通过其产物5-InsP7介导了DsbA-L/Ero1-Lα/脂联素/ERp44复合体的形成。
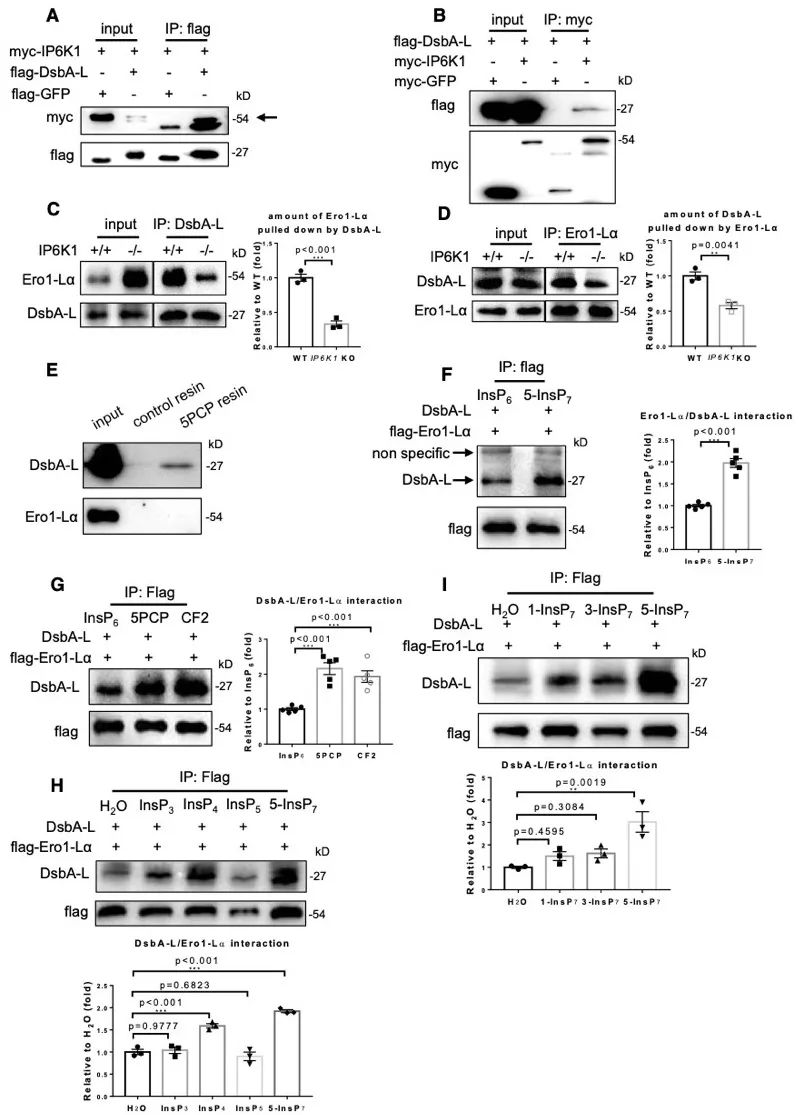
脂联素在脂肪细胞的合成存在明显的“浪费现象”,即50%的新合成的脂联素不被分泌入血而是在细胞内被降解。有意义的是,DsbA-L/Ero1-Lα/脂联素/ERp44复合体正是决定脂联素降解的关键因素。研究人员发现,通过干预IP6K1来抑制5-InsP7的表达量通过减少脂联素的降解来上调脂联素的蛋白水平。
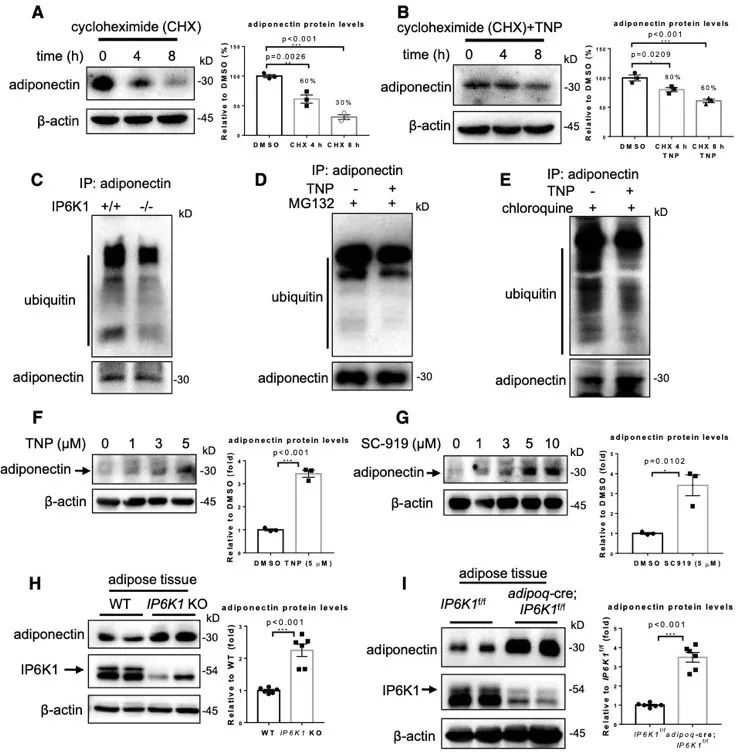
最后,作者利用IP6K1抑制剂验证了IP6K1作为干预靶点减轻心肌缺血再灌注损伤的有效性,并且利用脂联素敲除小鼠证实抑制5-InsP7通过脂联素来缓解心肌缺血再灌注损伤。
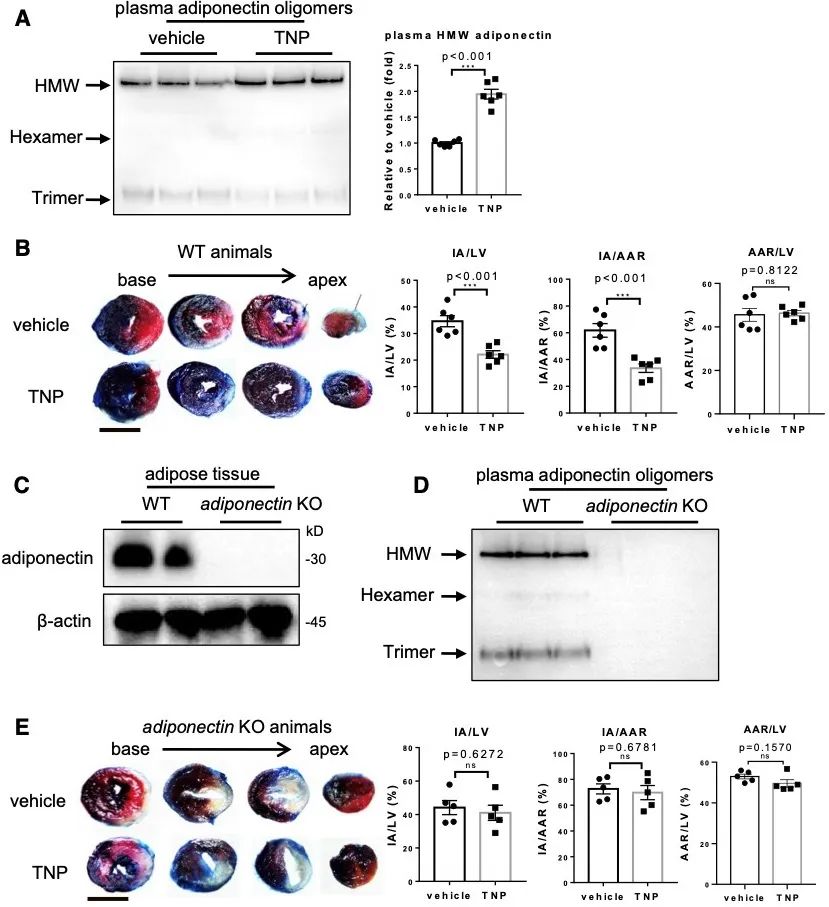
综上,本研究发现抑制5-InsP7的表达通过上调血浆脂联素来减轻心肌缺血再灌注损伤,并阐明了IP6K1通过其产物5-InsP7调控脂联素的分子机制。研究结果提示,IP6K1可能是潜在的心血管疾病的干预靶点。
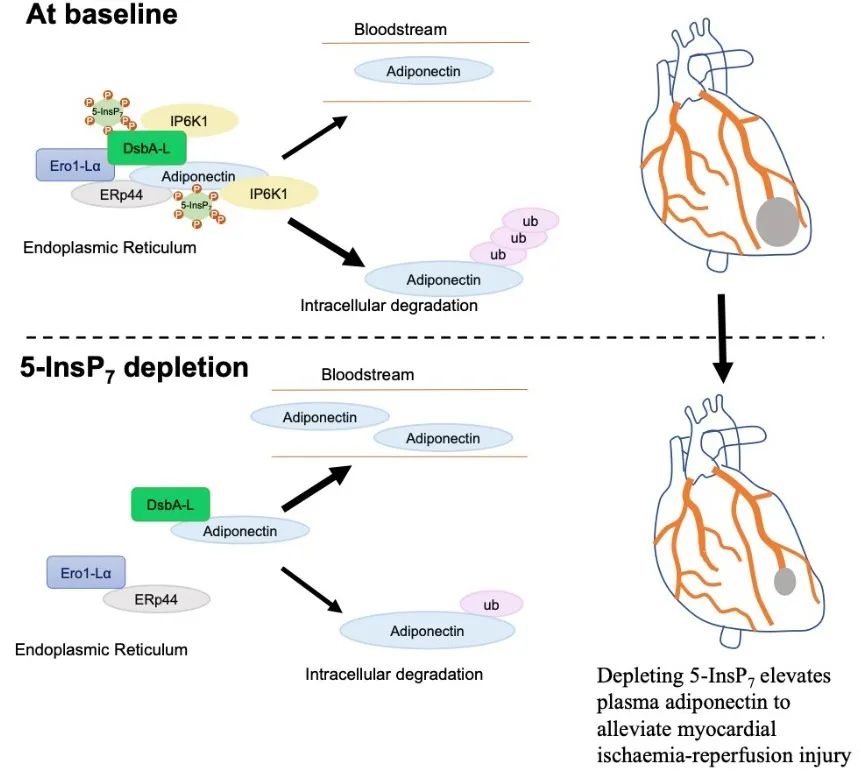
天津医科大学医学生理学专业付琳博士为本文第一作者,天津医科大学朱毅教授和上海交通大学医学院附属新华医院付成来研究员为本文通讯作者。课题得到了牛津大学Barry Potter教授、德国莱布尼茨研究所的Dorothea Fiedler教授和天津医科大学张栩研究员的帮助,感谢天津医科大学艾玎教授和刘彤教授的大力支持。本研究得到国家自然科学基金项目和上海市自然科学基金的资助。
原文链接:
https://doi.org/10.1093/cvr/cvae017
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








签到学习
80
#心肌缺血再灌注# #IP6K1#
86