
Psychiatry Res. :精神疾病患者的遗传多样性分析
2024-01-26 xiongjy MedSci原创 发表于上海

基因多样性是基因在进化过程中逐渐形成的固有属性。重度抑郁症、阿尔茨海默病、分裂情感障碍的基因多样性与 "正常 "多样性值有显著偏差。近三分之一的基因相互关联。
关于精神疾病的病因和发病机制的知识很少,无法可靠地预测特定患者是否以及何时会对特定治疗产生反应; 在个别情况下,几乎不可能做出任何可靠的预后。至于假定与精神疾病发病机制有关的遗传易感因素,证据显然反对单一原因,因为精神疾病在家庭中聚集,而且精神疾病不遵循简单的孟德尔遗传模式。在具有多个受影响受试者的家庭中未观察到同型诊断模式。通常,一级和二级亲属的临床诊断似乎与指示病例的主要诊断无关。
精神疾病的发病机制因病因异质性而进一步模糊,这表明多种途径可导致相同的临床表现。最可能的病因是多种遗传易感内源性因素和多种外源性因素之间的复杂相互作用。
这篇文章分析了遗传多样性的不规则性在多大程度上将精神病患者与健康对照组区分开来。

研究方法上,本文中的遗传多样性通过多维 "基因载体 "进行量化,"基因载体 "由位于 100 个候选基因中每个基因内的 4 至 8 个多态 SNPs 组成。每个基因所观察到的不同基因型的数量被称为该基因的 "多样性指数"。非线性神经网络 (NN) 通过“隐藏”层将输入层的“神经元”(受试者的基因载体)与输出层的“神经元”(受试者的精神病学诊断)连接起来。这篇文章目标是构建 NN 模型,通过基因载体根据精神病学诊断对所有 1698 名受试者进行正确分类。NN连接通过权重矩阵实现、模型拟合算法使权重空间中的误差函数最小化(“拟合优度”)。最流行的模型拟合策略是反向传播算法,它使用梯度下降方法寻找误差函数的最小值。模型的可实现精度主要取决于所包含的信息、基础数据的质量以及为模拟非线性相互作用而实现的中间层数量。
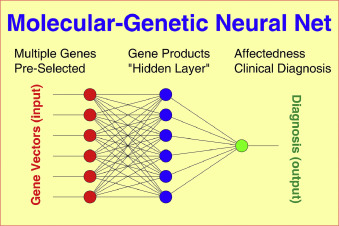
图 1.多层神经网络 (NN) 的主要模式
在1698名受试者的中欧人群中,所选候选基因的多样性指数范围为18(CYP2C19)至476(GPR39),平均值为109.4±82.8。由下图可以看到,多样性指数集合的分布呈现出两个峰值(多样性指数在70和170附近),以及7个基因表现出多样性指数在250以上。
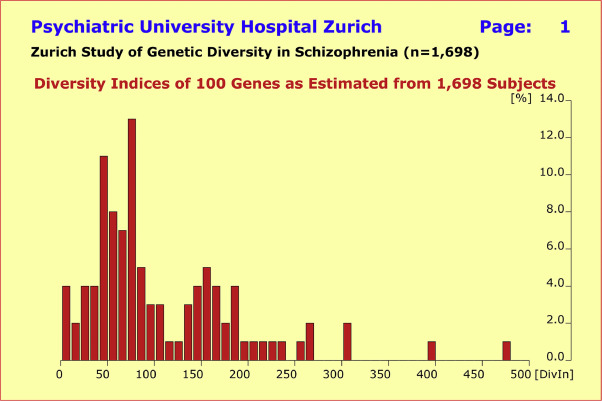
图2.在 1,698 名中欧受试者(包括少数美国人)中观察到的 100 个基因多样性指数的分布。
100*24 规范校准曲线,涵盖该项目的所有 100 个候选基因和群体大小,在散射方面表现出非常稳健的行为,如下图所示,两条曲线在形状和陡峭度方面的差异表明基因类型不同,CYP2J2属于左侧基因组(分布峰值在70左右),SLC6A6属于中间基因组(分布峰值在170左右)。

图3.多样性指数与样本量的函数关系,样本量范围为 50 至 1,700。
此外,基因型模式的分布显示健康对照组和 5 个诊断亚组的患者之间没有实质性差异。尽管单一基因型模式的比较偶尔会比Bonferroni校正达到统计学意义,但由此解释的表型方差非常小且非累加性。
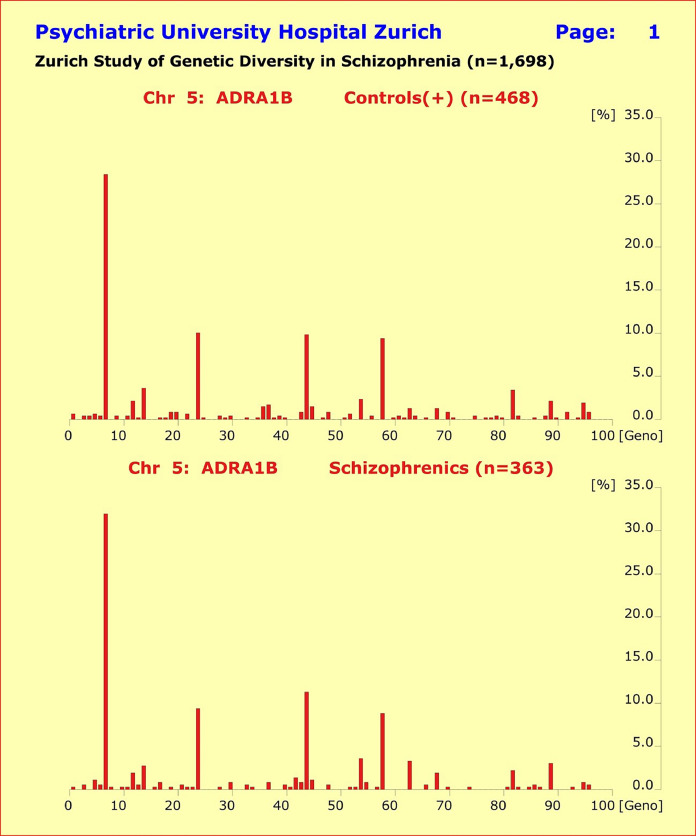
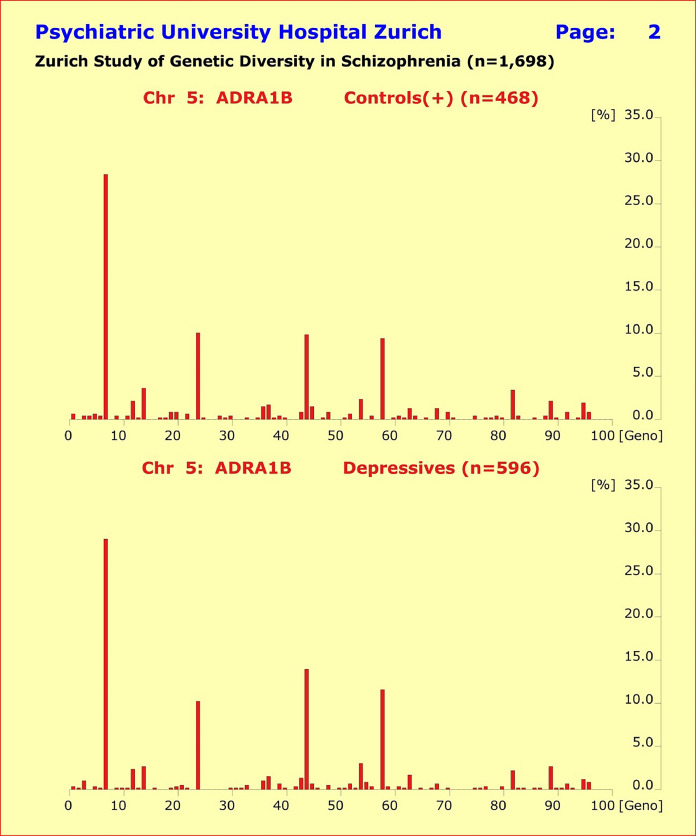
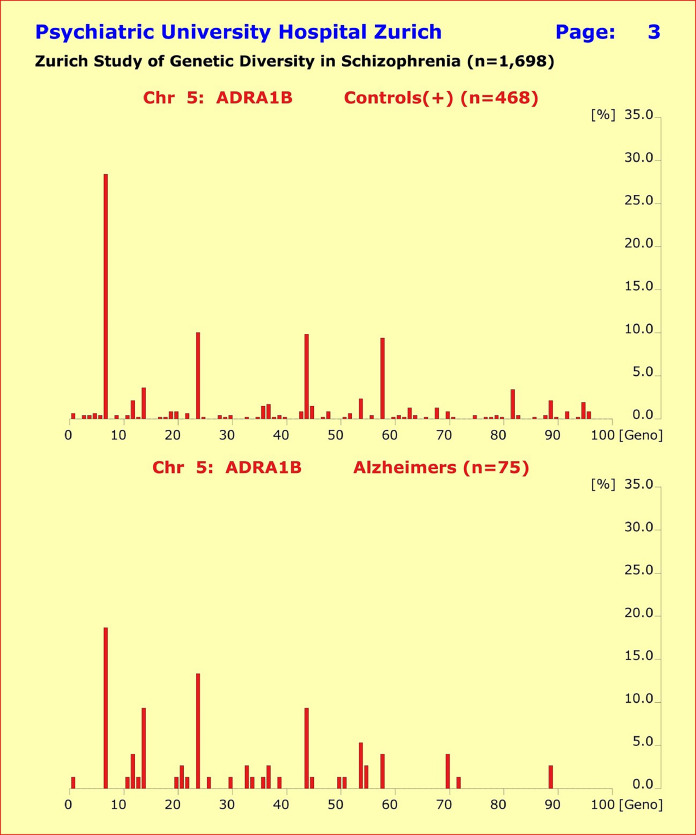
图 4 abc. 所研究基因的基因型模式分布显示,健康对照组(n=468,上半部分)与所研究的 4 个诊断亚组的患者之间没有实质性差异。
总之,从结果看,多样性指数与其组成的 SNPs 数量只有微弱的相关性(解释方差为 20.5%),这表明基因多样性是基因在进化过程中逐渐形成的固有属性。重度抑郁症、阿尔茨海默病、分裂情感障碍的基因多样性与 "正常 "多样性值有显著偏差。近三分之一的基因相互关联,关联度从 0.0303 到 0.7245 不等。
这篇文章核心发现是 "奇异基因",这些基因具有独特的基因型模式,只出现在患者身上,而不出现在健康对照组中。神经网络产生的非线性分类器能正确识别多达 90% 的患者。诊断亚组之间在基因型水平上的重叠表明,诊断交叉脆弱性很可能涉及主要精神障碍的发病机制,且临床定义的诊断可能并不构成病因实体。总之,通过对基因中基因型模式的变化以及基因之间相关性的详细分析,非线性分类器能够在基因型水平上非常稳健地将精神病患者与健康对照组区分开来。
原始出处:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













通过对基因中基因型模式的变化以及基因之间相关性的详细分析,非线性分类器能够在基因型水平上非常稳健地将精神病患者与健康对照组区分开来。
94
#遗传学# #基因组学# #精神疾病#
91