
【Blood Rev】特发性多中心型Castleman病的诊断和治疗新进展
2023-12-17 聊聊血液 聊聊血液 发表于上海

由于过去十年中实验室检查和药物治疗取得重大进展,Evan Lang和Frits van Rhee两位教授针对 iMCD 的新发展和报道进行了更新审查并撰写综述。
Castleman病
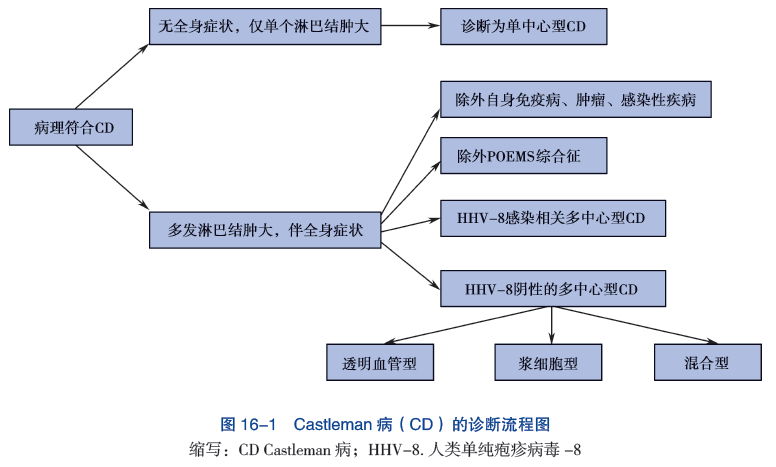
Castleman病于1954年首次报道,根据淋巴结分布和器官受累部位的情况不同,可分为单中心型Castleman病(UCD)和多中心型Castleman病(MCD)。前者发病率相对较高,仅累及单个淋巴结区域,全身症状反应较轻,主要治疗方法为手术,预后较好;后者发病率相对较低,累及多个淋巴结区域,多有全身症状、血细胞减少,甚至威胁生命的脏器功能受损,预后较差。
MCD可根据病因进一步细分:人类疱疹病毒8型 (HHV8) 相关MCD(HHV8-MCD);POEMS综合征相关MCD (POEMS-MCD);特发性MCD(iMCD),也称为 HHV8 阴性MCD。特发性 MCD 可通过临床表型进一步分类:重度iMCD表现为一系列异常实验室检查和临床结果(血小板减少、全身水肿/腹水、发热、骨髓网硬蛋白纤维化或肾功能不全和器官肿大(肝肿大和脾肿大),即TAFRO综合征(iMCD-TAFRO),另一种通常较轻的表型为非特指型(NOS;iMCD-NOS)。
由于过去十年中实验室检查和药物治疗取得重大进展,Evan Lang和Frits van Rhee两位教授针对 iMCD 的新发展和报道进行了更新审查并撰写综述,近日发表于《Blood Reviews》。

实践要点
•iMCD的诊断可能具有挑战性,但如果无法确定淋巴增生性疾病的其他原因,如恶性肿瘤或感染,则应怀疑iMCD。
•诊断标准(NCCN指南和国际共识标准)现已可用,应遵循。
•与不同专业的合作是患者治疗的最佳选择。
•血液学和肿瘤学在 iMCD 管理中发挥重要作用。
流行病学
多中心型CD 是一种罕见疾病,特发性 MCD 占所有 MCD 病例的1/2,在美国每年约有1100例新发病例。在2017年引入 CD 的疾病特异性国际疾病分类 (ICD) 代码和发表 iMCD 的国际循证诊断标准之前,难以获得准确的人群估计值且存在差异。此后基于索赔的分析估计,2017年和2018年 iMCD 的发病率分别为3.4例/100万和3.1例/100万。据估计,2017年和2018年 iMCD 的患病率分别为6.9例/百万人和9.7例/百万人。
iMCD 的预后历史上一直较差。一项回顾性分析跨越IL-6 拮抗剂司妥昔单抗获批前的60年时间,其中35%的 MCD 患者在诊断后5年内死亡。特发性 MCD 也会导致显著的发病率。在最近使用 CD 特定 ICD-10 代码和 iMCD 循证诊断标准的人群水平美国健康索赔分析中,在诊断后的第一年,约半数的患者至少住院1天,近43%的患者至少有1次急诊室就诊,常见的就诊原因为血栓栓塞、肾功能不全和呼吸问题。此外,与非 iMCD 的匹配对照组相比,更多 iMCD 患者发生骨髓恶性肿瘤 (10.0% vs 0.6%) 和实体恶性肿瘤 (18.1% vs 7.4%),可能是由于白细胞介素-6(IL6) 诱导的整体免疫抑制。
发病机制
对iMCD发病机制的认识不如其他类型MCD。IL-6 已被确定为 iMCD 疾病过程的主要介质,作为一种多功能细胞因子,IL-6可调节 B 淋巴细胞和浆细胞的生长和分化、免疫反应和急性期反应,对炎症和造血也有多效性作用。IL-6和其他细胞因子可驱动 iMCD 中发生的多中心淋巴结病、临床症状和炎症实验室标志物(如 ESR 和CRP)。更严重的 iMCD-TAFRO 患者还可出现细胞因子风暴,可诱发多器官功能衰竭,甚至死亡。
大阪课题组(1986年)和Yamasaki 等(1989年)分别克隆了白细胞介素-6及其受体。1989年也首次报道了 IL-6 与 CD 的相关性。
IL-6 表达失调可诱导 Castleman 样综合征,随后发现抗 IL-6 受体抗体可抑制 IL-6 转基因小鼠的 CD 样症状。1994年首次报道CD 患者在长期使用鼠单克隆抗 IL-6 抗体后疾病体征和症状迅速消退;异常实验室检查值也在数天内改善,但在停止治疗后复发。虽然 IL-6是 iMCD 最常见的病理驱动因素,但 iMCD 的确切病因仍有待完全阐明,IL-6和其他细胞因子生成细胞的来源也有待完全阐明。近期一项全面研究排除了 iMCD 的病毒原因。IL-6 存在于不同的亚型、单体、二聚体和三聚体中,可与伴侣蛋白复合。需要强调的是,患者接受抗 IL-6 治疗后不应测量IL-6,因为目前的技术无法区分结合和未结合的IL-6;通常会导致 IL-6 值人为升高,不应用于指导治疗决策。CRP 是 IL-6 活性的替代测量指标,可应用于诊断和监测。
iMCD的诊断
2018年,基于HHV8阴性/iMCD,首次建立专家共识诊断标准,并被NCCN指南所参考。该标准包括2个主要标准和11个次要标准,以及排除可模拟 iMCD 的疾病(表1)。主要标准包括特征性淋巴结组织病理学和多中心型淋巴结肿大,次要标准则细分为临床特征和实验室检查异常。

iMCD 的诊断需要医生和病理学家的积极合作。病理结果本身不足以做出 iMCD 的诊断,因为其他疾病可能引起类似的淋巴结异常(图1)。需要切除活检来确定与 iMCD 改变相一致的组织病理学淋巴结特征(主要标准),而细针抽吸和组织芯活检不太可能为识别 iMCD 的组织病理学特征提供足够材料。淋巴结样本的血液病理学检查包括基于5个特征的分级:生发中心 (GC) 退化、滤泡树突状细胞 (FDC) 突出、血管分布、浆细胞增多和GC增生。一致和可靠地确定组织病理学特征需要相当多的专业知识。一项2期研究中患者淋巴结样本的亚组分配分析显示,由3个独立的血液病理学家专家组(当地、中心和专家组)进行审查,仅23%(18/79) 的患者的亚型分配一致;但iMCD的确切组织病理学亚型对治疗无影响。
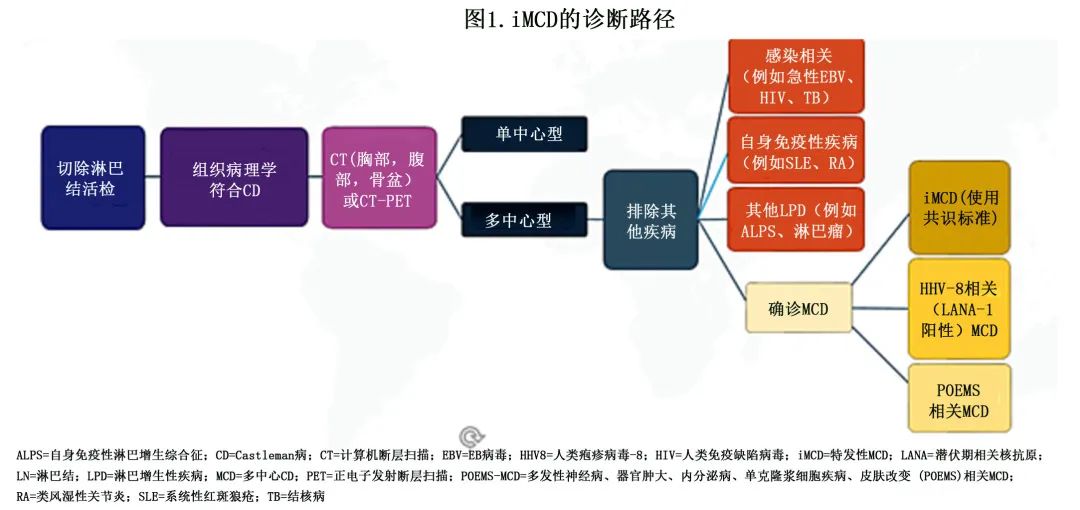
由于组织病理学至关重要,包括血液学家和其他专业的专家多学科团队应纳入经验丰富的病理学家。
严重程度
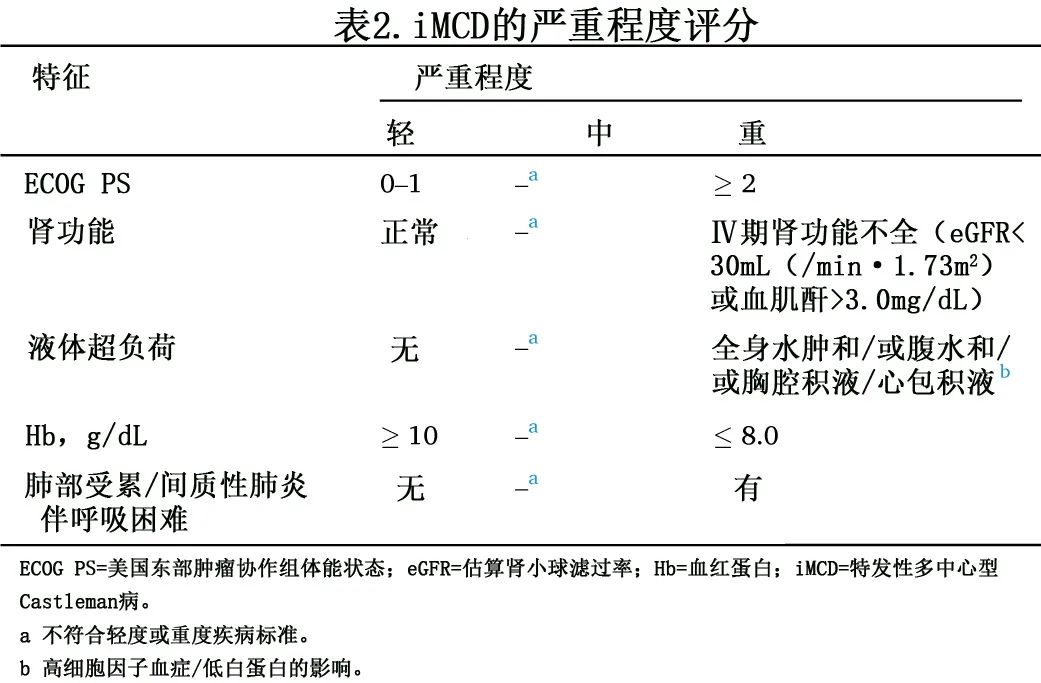
iMCD 的严重程度可能因临床表现不同而存在很大差异,从轻度症状到危及生命的器官衰竭不等。根据专家意见和证据审查而制定了疾病严重程度评估的简单标准(表2)。重度疾病需符合≥2条重度标准,中度iMCD 病例为不符合轻度或重度疾病的标准;在选择治疗时必须考虑疾病的严重程度。
亚型
可根据2种表型描述特发性 MCD 临床亚型:TAFRO- iMCD与NOS-iMCD。iMCD-TAFRO 的诊断标准见表3。iMCD-TAFRO患者表现出与 iMCD-NOS 患者不同的细胞因子谱,IL-6升高不太明显,VEGF增加。与 iMCD-NOS 相反,iMCD-TAFRO患者通常出现血小板减少和碱性磷酸酶升高,而血清丙种球蛋白水平正常或轻度升高。iMCD-TAFRO 患者的淋巴结通常血管丰富,病理学通常被称为高血管而非透明血管。

对于怀疑 iMCD-TAFRO 的患者,应评估血管内凝血、纤维蛋白溶解、骨髓网硬蛋白纤维化和巨核细胞增生。iMCD-TAFRO 的鉴别诊断包括重度全身性炎症性疾病,如巨噬细胞活化综合征和噬血细胞性淋巴组织细胞增生症 (HLH)。而iMCD-NOS倾向于比 iMCD-TAFRO 侵袭性更低,且临床病程各不相同,不符合 iMCD-TAFRO 标准的 iMCD 病例归为iMCD-NOS。iMCD-NOS的特征为血小板增多、γ球蛋白水平升高、全身水肿频率降低、碱性磷酸盐水平降低和对糖皮质激素治疗的反应性增加,但后者不应作为唯一治疗。
即使已建立诊断标准,iMCD的诊断仍极具挑战,其原因很多,包括与其他疾病重叠的临床和组织病理学特征、临床表现的异质性以及缺乏特异性生物标志物。特发性 MCD 可酷似其他疾病,如淋巴瘤和感染性、恶性和自身免疫性疾病。在 iMCD 患者的回顾性数据库索赔分析中,约89%出现流感样症状(即全身或 B 症状),58%出现液体积聚,34%出现肾功能不全,24%出现肝脾肿大,19%出现血小板减少,这可能导致患者就诊于可能不熟悉该疾病的专科医生(例如传染病科、风湿科、肾病科、耳鼻喉科和全科)。此外,没有指示 iMCD 的特异性生物标志物。对于出现淋巴结肿大、全身 B 症状和/或多器官系统功能障碍的患者,鉴别诊断中应包括 iMCD。
治疗指南
2期数据有助于建立治疗指南(图3),指南旨在减少从就诊到治疗的时间,并可能改善临床结局和生存率。司妥昔单抗是很多国家唯一获批用于治疗 HIV 和 HHV8 阴性 iMCD 的药物, NCCN 指南将其列为首选一线治疗选择。在司妥昔单抗不可用或治疗方案(每3周一次IV 输注)不耐受的情况下,患者和临床医生可选择其他治疗,包括托珠单抗(抗 IL-6 受体单抗)联合或不联合糖皮质激素。司妥昔单抗和托珠单抗均通过阻断 IL-6 的下游效应发挥作用,IL-6是 iMCD 的已知驱动因子。抗 IL-6 治疗无效患者的替代治疗包括利妥昔单抗和/或化疗。

如果使用利妥昔单抗联合或不联合化疗作为一线治疗,司妥昔单抗应视为二线治疗,治疗应持续至疾病进展。对于复发/难治性 iMCD 患者,治疗选择包括联合化疗(CHOP、CVAD、CVP或多柔比星脂质体)联合或不联合利妥昔单抗或其他药物,如蛋白酶体抑制剂(硼替佐米)和免疫调节剂。
司妥昔单抗也应考虑用于治疗患有炎症综合征的不可切除的症状性 UCD 患者。虽然支持司妥昔单抗在这种情况下疗效的数据有限,但国际循证治疗指南建议在考虑广泛手术或放疗前 CRP/ESR 升高的不可切除、症状性 UCD 患者使用抗 IL-6 治疗(包括司妥昔单抗)。
总结和未来方向
iMCD是一种罕见疾病,由于其症状和实验室检查结果的异质性,可能诊断不足。iMCD 的确切病因目前尚未完全阐明,但可引起显著的发病率和死亡率。在过去十年中对 iMCD 的理解取得了重大进展,包括诊断标准、治疗指南和 FDA 批准的有效药物 (司妥昔单抗)。对司妥昔单抗无应答或无法获得司妥昔单抗的患者,其他治疗选择包括托珠单抗、利妥昔单抗、化疗、蛋白酶体抑制剂或免疫调节剂如西罗莫司,然而对于司妥昔单抗无应答的患者,仍存在存在未满足的需求。业内在提高对这种罕见疾病的认识方面取得了巨大的进展,帮助医疗专业人员识别该疾病并为患者提供最佳治疗。还需要进行研究来了解该疾病的病因和发病机制,也就是可能触发 iMCD 的环境和个体因素,以及它们如何相互作用。此外,免疫失调在 iMCD 中的作用以及导致治疗耐药和复发的机制仍未解决。
参考文献
Lang E,Rhee FV.Idiopathic multicentric Castleman disease: An update in diagnosis and treatment advances.Blood Rev . 2023 Dec 5:101161. doi: 10.1016/j.blre.2023.101161.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







学习了,谢谢分享
81
不错,学习了
63
Castleman病于1954年首次报道
53
#Castleman病# #特发性MCD#
63