对比中国和美国医药的投资回报率,中国在20%以上,美国只有3%,目前中国的风险没有美国高。
原因是美国创新药多,失败率高;而国内企业的临床试验,一方面是大多数还处于较早期的I期、II期临床阶段,进入III期较少,另一方面这些品类很多都是me-too或me-better,这类产品的临床成功率要高得多。
临床试验是一个成功率很低的活动
新药临床试验是一个成功率很低的活动,药物从I期临床试验开始要花很多的精力和资源,因此,临床试验失败的成本非常高。
根据数据统计,临床I、II、III期的成功率分别为63.2%、30.7%和58.1%,从下图可见,临床I、II、III期的成功率呈“V”型转折,II期的失败率远远高于I期和III期。
总体看,从I期到上市,临床研究的成功率不到10%,其中肿瘤药的成功率更低。
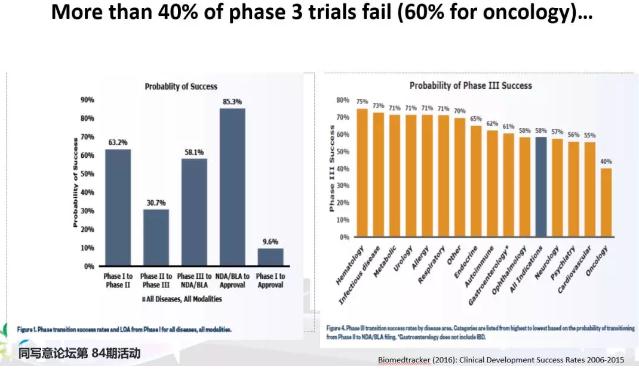
图1:各阶段临床试验成功率 & 不同适应症III期成功率
临床试验失败的主要原因
在不同临床研究阶段,临床试验失败的原因有所不同。
有文献对160个临床I期失败案例和89个临床II期失败案例的原因,从安全性、有效性以及公司策略等方面进行了分析。
发现临床I期失败最多的原因是安全性,因有效性失败的比例不足9%(14/157);到临床II期,失败最多的原因是有效性,比例达35%(31/89);而到了临床III期,有效性作为最主要的失败原因,占比高达55%。

图2:各阶段临床研究失败原因小结
而纵观整个临床研究,失败的主要原因有:
药物无效(无法控制)。
并不是每一个产品都会有预期的作用或者副作用。
临床试验设计问题(可以控制)。
如何根据资料、文献设计临床试验,减少临床试验失败风险成为主要课题。
操作问题(可以避免)。
临床试验人员的责任就是要把好药做成功。如果临床试验设计很合理,药物也很好,做失败了,那么这是做临床试验人员的失职,操作问题是我认为最应该避免的错误。
决策问题(决策很重要)。
公司在临床I期或者II期失败时候,如何评估失败产品?有没有足够的信息去评估(大多数情况下是没有完整的数据而必须做出决定)?如何决定是不是要继续投资?……
这些问题,其实是通过经验以及对资料的分析,对竞争格局的判断,还有公司内部策略等因素来进行决策的。因此,决策很重要。
Case study
怎样通过合理决策,避免III期临床失败?
案例1:辉瑞公司适应症为肾细胞癌的产品“舒尼替尼”(商品名:索坦)
舒尼替尼曾经做了很多临床试验,其中有七、八个III期临床试验失败。例如,乳腺癌的临床试验。II期临床的结果,舒尼替尼单臂临床实验应答率11%,而卡培他滨(商品名:希罗达)在同样的病人应答率为20%。但辉瑞决定继续开展这一临床试验的III期。
最终试验结果:舒尼替尼的无进展生存期(PFS)比对照组卡培他滨的短了一半,客观缓解率11%,而卡培他滨有16%。
回顾II期临床数据,这个III期失败完全可以被提前预测:拿一个本来就已经比对照组差的资料和数据,去决定继续进行III期临床研究,这有点像抱着侥幸心理——“万一成功了呢”?
通过这个案例可见,要尊重数据。这很重要。
案例2:辉瑞的一个IGF-1抑制剂:Figitumumab
II期临床试验结果显示:低剂量组10mg比对照组还差,高剂量组20mg比对照组稍好一点。
但如果调整7%、11%、46%和52%的crossover time,与对照组没有区别,即用药以后crossover变差,这是一个信号。
但是Sponsor认为还可以,因为高剂量组20mg hazard ratio从 0.8下降到0.56,结果变好。是不是还可以继续试验?
于是开展了两个大的III期临床试验,分别是:
“Figitumumab与厄洛替尼联用”
“Figitumumab与化疗药物联用”
这个临床试验还没做完就发现结果是阴性,被暂停,试验结果显示用药后试验组比对照组差,临床试验失败。这一项研究辉瑞在2009年发表了文章。
但是后来对临床试验数据进行数据梳理和确认发现:2009年数据有误,缓解率从78%下降到50%多,客观缓解率从54%变成37%,这说明当时没有充分确认,导致对结果错误报道,于是文章在2012年被撤回。
此外,即使当时特别看好的鳞癌中79%的应答率,也修正为后来的42%。改正前后的资料差异很大。由此做出做III期临床的决策,结果出来非常差。
案例3:IDO抑制剂
IDO临床一期和二期的结果显示55%的应答率。参照PD-1单一疗法约30%左右的应答率,他们认为从30到55增加了很多,这个结果看起来不错,但这是单臂、联用的临床试验。
然后,业界做了一大批III期临床试验,例如大公司BMS、Incyte就做了好几个临床。
今年6月份ASCO报道了该类临床试验结果:PFS的曲线用药组与对照组一模一样,OS也一模一样——试验的质量、结果都很好很可靠,但可惜IDO抑制剂是无效的。
案例4:Motesanib(AMG-706)
Motesanib是一种小分子多靶点酪氨酸激酶抑制剂(MTKI),是我当时领导开发的一个产品。
首先开展了一个180例甲状腺癌的II期单臂的临床试验。甲状腺癌有两种,甲状腺癌有两种,一种是MTC。(髓样甲状腺癌),一种是DTC(分化型甲状腺癌)。
试验结果对两种不同的甲状腺癌的组织学表现很好,肿瘤全部缩小。
这是一个较大的II期单臂临床试验,但与FDA沟通中,FDA认为这是单臂试验,因为没有对照组,无法判断药是不是有效,建议做一个随机的对照试验——但是这就要耽误很多年。
而后公司继续做其他II期试验,一个是乳腺癌,一个是非小细胞肺癌。乳腺癌试验有安慰剂和Avastin(bevacizumab)两个对照。
结果Motesanib和安慰剂早期有区别,晚期没有区别。试验结果比较差。在非小细胞肺癌中,是跟Avastin对比,试验结果跟Avastin差不多或者略差。
最后公司需要要做决定,下一步怎么做?甲状腺癌是否重新做,试验的成功率如何?并考虑到市场规模,竞争者等因素,我们认为:
虽然甲状腺癌试验结果很好,如果做临床试验基本上肯定成功,但是当时计算美国甲状腺癌市场规模只有5000万美元,5000万美元对于安进来说是太小的数字,根本不想做(不到5亿美元的市场规模大公司一般不大愿意去做这种产品)。
GBM(多形性胶质母细胞瘤)也可能成功,但也是很小的市场规模。
Sarcoma(肉瘤)也有很多资料证明这个产品可以成功,但是市场也很小。
乳腺癌、肺癌,都有很大的市场规模。但II期试验结果乳腺结果比较差,肯定不能做。肺癌结果虽然并不突出,但还是有点效果,跟Avastin差的可能不是很多。
于是,最后决定继续非小细胞肺癌试验。
于是,我们开展了一个III期临床试验,在30个国家150个sites,入组1500个病人。
结果如图,左边是无进展生存期PFS,右边是OS。PFS0.79的hazard ratio,OS是0.9,没有统计学显着差异,做了五年的临床试验宣告失败。
随后,对亚洲人亚组数据进行分析,结果发现亚洲病人结果特别好:非亚洲人是1.0的hazard ratio,没有差异;但是如果是亚洲人,PFS从对照组14.5个月到给药组20几个月,5个月的差异在非小细胞肺癌是一个巨大差异,而且0.669的Harzard ratio。
这个药在亚洲病人亚组的表现似乎很不错。并且我们做了各种各样的基线调整、治疗后区别,结果令人信服。
武田制药认为这很好,于是安进将产品授权武田,在日本继续做另一个三期临床试验。结果PFS 0.8,OS 0.9,跟之前全球研究一模一样的结果,试验失败了。
从这个临床试验得到的经验教训:
要尽早跟注册人员去沟通,确保你的策略能够注册。
Amgen最开始没有跟FDA讨论甲状腺癌的注册策略,因此导致单臂的II期临床试验虽然结果很好,但对于注册来说是无效的。
选择适应症一定不单单考虑市场,要看胜算多少,PTRS很重要。
例如甲状腺癌虽然适应症很小,市场规模不大,但有一个适应症能上市,有销售收入然后再做其他的话,比一个适应症都没批就失败要好得多。
对临床试验亚组的分析要非常非常的谨慎。
因为亚组是一种“假设成立”,是去衍生发展一个潜在的假设,因此基于产品的亚组分析一定要去确认是不是真的,确认的方法就是依据随机、对照临床试验。我认为这是很重要的一个教训。
Case study
产品的开发策略也很关键
案例5:Regorafenib
上述案例4的产品Motesanbi其实跟这些索坦很相似,都是anti-VEGF。
当时所有的anti-VEGF的小分子药全做结直肠癌,一线治疗全失败,包括Sutent、Nexavar、Recentin、Motesanib,他们做fisrt line跟化疗联用的一线治疗,临床试验III期全都失败。
但有一个成功的产品是拜耳的瑞格非尼(Regorafenib)。为什么成功?这个产品做的一个三线的单药临床试验。
当时独树一帜,因为没有人考虑做一个单药三线临床试验。用的对照相当于就是安慰剂,没什么治疗作用。结果0.77的Hazard ratio,不算特别好,OS是从5个月延长到6.4个月。
由此可见,产品开发策略也很关键。
案例6:Rilotumumab
这是Amgen的另一个产品Rilotumumab,虽然我不是主要负责,但是也介入一些开发工作。
II期临床试验结果一般。右边Over Survival曲线,有点区别,但不是很大,0.7几的Hazard Ratio,这时Amgen公司已经宣布把这个项目终结。
结果做完试验,课题组做了一个样本分析,又是subset分析。如果看cMET高的病人,结果差别大,OS 0.48 Hazard ratio,看Median的差不多是2倍,很好的结果。
根据subset分析,Amgen开展一个全球多中心的,600例病人的胃癌一线III期临床试验。
试验尚未做完,中期分析就显示试验失败了,不但失败,对照组比给药组明显更好,1.36的Hazard ratio,增加36%的死亡风险。
最后结论,用Rilotumumab对胃癌的OS没有影响且有风险。
Amgen宣布结束Rilotumumab的临床试验。之前已经终结过的项目,花这么多钱以后又重新终止,即通过一个小样本的亚组分析来主导一个III期临床试验设计非常危险。
所以subst分析可能会误导、可能会非常危险!
案例7:Ramucirumab
但是,基于Subst分析的III期临床试验有没有成功的呢?也有。像礼来的产品Ramucirumab。
Ramucirumab是一个anti-VEGF2的抗体,做了一个肝癌的II期临床试验,结果0.86 的Hazard ratio,有一点点区别但不是很多。
Subset分析发现如果病人baseline的AFP大于等于400ng/mL,结果看起来不错,0.67 Hazard ratio。
礼来基于此开展了二线肝癌的III期临床试验。III期临床试验结果跟II期差不多,0.67 Hazard ratio,临床试验成功,获得美国FDA肝癌二线治疗的批准。
这个Subset比较大,而且它是预先设定的,这是Subst分析很重要的两点。
如何减少III期临床失败的风险?
减少III期临床风险这个任务有时候是可能的,有时候是不可能的。
因为很多时候小样本的II期临床试验并不能让我们了解一个分子的全部特性,所以失败是肯定的。
但我们的责任是要减少,最小化那些不应该失败的错误。
详细深入地了解研究资料,靶点,早期数据,安全性等,非常重要。
要合理开展I期、II期早期临床试验。
众多的IDO III期临床试验,没有一个II期是随机的,全是新单臂联合试验。
联合试验如果是II期单臂,很危险,因为产生结果的原因并不清楚,联合用药时,是不是里面每一个药都有用——这在结果从10%增加到90%时容易判断,但如果如果结果是从40%增加到60%,则不容易判断。
客观仔细分析所有可获得的数据很重要。
看自己做的项目就像看自己的宝宝,可能不完美,只看好的不看坏的。因此,不要忽略不好的数据,不能只看高剂量结果很好,而不去问低剂量组为什么不好?
科学永远是对的,但我们不会知道全部。例如,为什么一个产品在高剂量有效,在低剂量不但无效而且更差,这是个很大的警示信号,一定要注意这样的数据。
还要有robust hypothesis,不能想当然或者存在侥幸心理。
Smart设计也很重要。
病人的选择,选什么样的临床试验设计,我觉得simple design is the best,越复杂越麻烦,例如A与 B对比,或者是“A与其他合用”与A对比,都是比较好的简单设计。
注意安全性,这也是临床试验失败的很重要原因。
注意地域性差别。
中国国内可能有这种差别,中国东北和南方区别可能也有一些,但不是很大。美国或者全球的区别会很大。有时候会因为一个区域造成临床试验失败的例子也很多。
高效、高质量开展也是减少III期临床失败很重要的因素。
运气也是成功因素之一,只是我们不能控制而已。
新药研发失败率很高,有时候真的需要一点好运气,像K药差点被毙掉。
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言