
Genome Biology:稳健、快速、低成本!INSERT-seq方法可对DNA整合特征进行高分辨率定位
2022-11-28 测序中国 测序中国 发表于安徽省

综上所述,INSERT-seq将基于扩增的富集、UMI扩增校正以及处理整合位点的计算框架相结合。
基因编辑新方法的发展使人类基因治疗(HGT)应用得以扩大发展。许多HGT策略都是通过添加DNA有效载体将治疗基因传送到受体中,以弥补遗传缺陷或为受体细胞提供合成功能。传统上,治疗性基因的传递是基于病毒和非病毒载体的,但其在基因组的整合位点不受控制,可能会给受体带来负面影响。
近年来,基于ZFN、TALENs和CRISPR-Cas9等工具的精准递送策略已实现快速发展,相对于传统的基于慢病毒(LV)载体、 rAAV载体和PiggyBac转座酶等技术而言,其可以在一定程度上避免意外的基因破坏或激活。在不受控制且精确的基因递送策略中,了解整合事件的综合特征对于评估安全性和精确性至关重要。因此,进入基因治疗时代,人们必须全面、综合地了解和评估这些治疗“载体”的精准性及其能否整合在基因组上的插入位点。
近日,西班牙巴塞罗那庞培法布拉大学的科研人员在Genome Biology上发表了题为“INSERT-seq enables high-resolution mapping of genomically integrated DNA using Nanopore sequencing”的文章。研究团队开发了一种名为“INSERT-seq”的测序方法,其主要工作流程包括single-tail adapter/tag(STAT-PCR)文库制备和牛津纳米孔(Oxford Nanopore)长读长测序。INSERT-seq方法将整合DNA的靶向扩增、基于唯一分子标记(UMI)的PCR偏差校正以及Oxford Nanopore长读长测序相结合,可以稳健、快速和低成本的方式解析已编辑的基因组中未知的有效载体整合位点,定量分析不同体外、体内样本的DNA整合特征,检测极限为1%。

文章发表在Genome Biology上
主要研究内容
增加reads长度提高对基因组插入事件的分辨率
为确定增加reads长度可以提供的分辨率增益,研究团队生成了一个模拟数据集,增加了插入-基因组结合位点的reads长度。当reads大小从250bp增加到1000bp时,检测到的整体插入位点增加了3.9%(图1a)。研究团队观察到,被短读长测序跳过的重复区域的结合位点是基因组中最长的可移动基因元件(MGE);插入位点对reads长度具有明显的依赖性,当reads长度增加时,该方法的敏感性也会增加。此外,在反转录转座子LTR、LINE和SINE中,研究团队使用长读长测序分别检测到4、3和2个新的插入位点(图1b)。
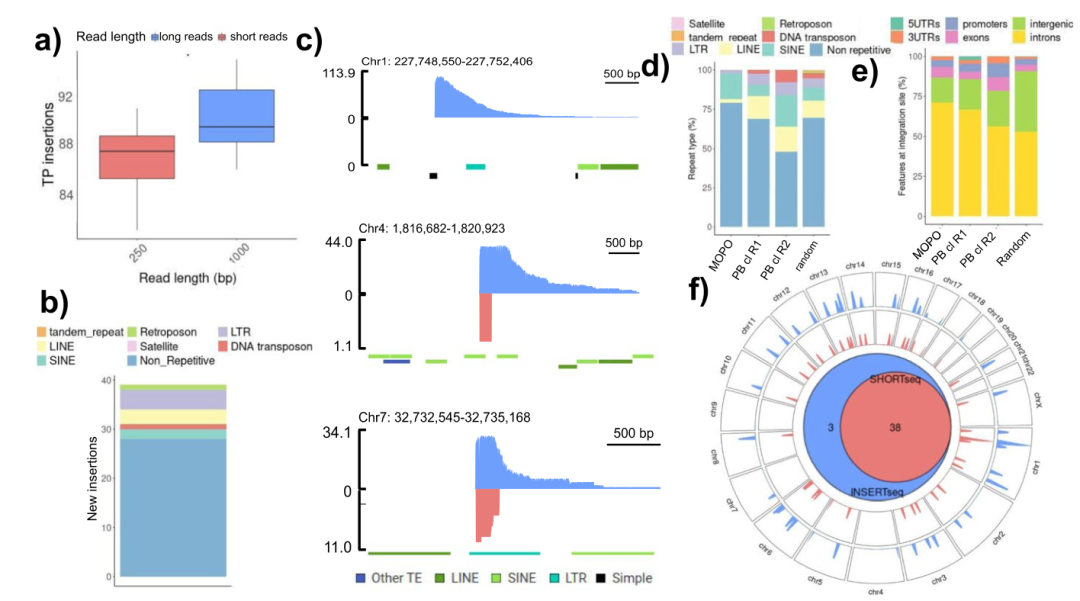
图1. read长度对解决插入部位的影响。来源:Genome Biology
接下来,研究团队在模型预测的最佳范围内使用嵌套的single-tail adapter/tag(STAT-PCR)与纳米孔测序耦合以获得长读长插入连接捕获(图2a),并在含有多个慢病毒插入的克隆扩增等基因细胞系(MOPO样本)上,将基于纳米孔的INSERT-seq与基于短读长测序的平台进行比较。结果显示,INSERT-seq检测到3个新插入(7.3%),其分别位于chr1、chr4、chr7处。
上述分析的MOPO样品中,在重复区域中发现的插入包括LINE、SINE、LTR等反转录转座子和DNA转座子(图1d),并且插入优先发生在内含子区域,其百分比高于随机分布的模型样本(图1e)。载体首选区域中重复元件丰度的变化可能会导致插入位点的整体检测结果不同,使得每个载体在解析位点选择方面的reads长度影响是可变的(图1d)。

图2. 优化的INSERTseq协议的实施。来源:Genome Biology
为确定INSERT-seq检测限(LOD),研究团队将一个在chr12位点有靶向插入的单克隆细胞系(MN2)和一个在chr6位点有插入的不同单克隆细胞系(MN7)进行恒定量的稀释,稀释倍数分别为1:1、1:10、1:100、1:1000和1:10000。在稀释度为1:100时检测到目标整合位点,从而确定INSERT-seq检测限为1%。
探究rAAV-8在小鼠肝脏中的全基因组整合和Cas9-PiggyBac嵌合转座酶的整合效果
为评估体内模型中INSERT-seq的性能,研究团队分析了重组腺相关病毒(rAAV)血清型8的小鼠的肝组织,以确定rAAV的全基因组整合图谱(图3a)。结果共检测到55次插入,其中有37次在两次重复之间是相同的(图3b);rAAV在内含子区域的优先插入(图3c),且插入位点与开放染色质区域相关(与ATAC-seq和DNAseI数据集相比)(图3d)。
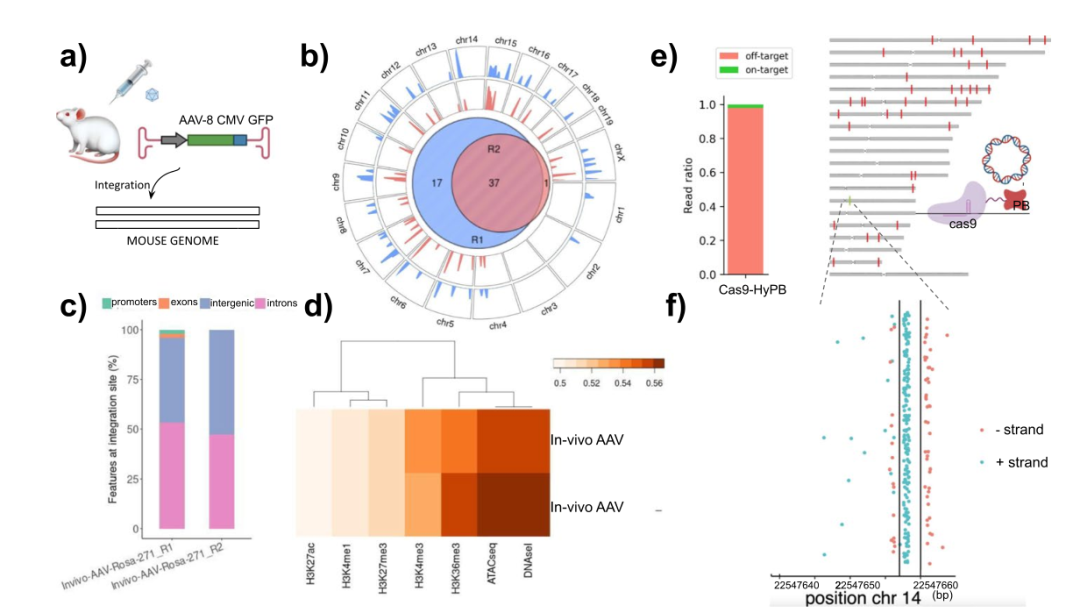
图3. INSERT-seq揭示了rAAV-8和PB基因组编辑框架中的整合模式。来源:Genome Biology
为评估INSERT-seq在分析新兴精确基因递送载体方面的潜力,研究团队确定了先前报道的Cas9-PiggyBac嵌合融合体的整合位点。通过INSERT-seq分析,研究团队在Cas9诱导的TRAC位点双链断裂处检测到了插入,与总整合量相比,其插入量很低,脱靶率为98%(图3e, f)。这可能是由于转座子转染的时间过长(约3周),特定位点对携带插入的克隆进行了正向选择。因此,在质粒侧翼区域添加PCR阻断寡聚物可能是提高新转染细胞插入位点覆盖率的一个解决方案。
总 结
综上所述,INSERT-seq将基于扩增的富集、UMI扩增校正以及处理整合位点的计算框架相结合。与传统的短读长测序方法相比,INSERT-seq能以更高的分辨率解析DNA整合位点,为确定重复区域的整合提供更高的准确性。研究团队已成功通过INSERT-seq方法分析了体外和体内样本基因组中的LV、转座子和rAAV DNA有效载体。此外,INSERT-seq还可用于捕获基因组中任何DNA有效载体的规范整合,将其与引物拼接结合,较高的检测灵敏度可以提高对基因组破坏事件和非规范整合结果的检测。
参考文献:
Ivančić, D., Mir-Pedrol, J., Jaraba-Wallace, J. et al. INSERT-seq enables high-resolution mapping of genomically integrated DNA using Nanopore sequencing. Genome Biol 23, 227 (2022). https://doi.org/10.1186/s13059-022-02778-9
·END ·
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




学习了
54