综述:格林-巴利综合征的病因,诊断,治疗与预后
2021-08-18 网络 网络

格林-巴利综合征(Guillain-Barré syndrome,GBS),又称吉兰-巴雷综合征,是急性弛缓性瘫痪常见的病因,以四肢的对称性无力、反射减退或消失为特征,病情常在4周之内达
格林-巴利综合征(Guillain-Barré syndrome,GBS),又称吉兰-巴雷综合征,是急性弛缓性瘫痪常见的病因,以四肢的对称性无力、反射减退或消失为特征,病情常在4周之内达到高峰(图1)。感觉症状,如感觉减退或麻木,常起始于远端肢体,呈对称性表现。是以周围神经和神经根的脱髓鞘病变及小血管炎性细胞浸润为病理特点的自身免疫性周围神经病,经典型的GBS称为急性炎症性脱髓鞘性多发性神经病(AIDP),临床表现为急性对称性弛缓性肢体瘫痪。
GBS最常见的亚型为急性炎性脱髓鞘多神经根神经病变(AIDP)和急性运动性轴索神经病(AMAN);其次为Miller Fisher 综合征(MFS),以眼肌麻痹、共济失调及深部肌腱反射消失为特征。总体来说,GBS的临床病程、严重程度和结局具有高度各异性。
GBS常发生在感染性疾病之后,前驱感染时免疫反应产生的抗体可与神经细胞膜上的神经节苷脂发生交叉反应,该自发免疫反应可引起神经损害或神经传导的功能性阻滞。前驱感染的类型和抗神经节苷脂抗体的特异性在很大程度上决定了GBS的亚型和临床病程。
在过去10年中,已有大量关于前驱感染和抗神经节苷脂抗体在GBS免疫病理过程中作用的新知识涌现。现在我们了解到前驱感染最常见的病原体为空肠弯曲菌,与GBS的AMAN亚型有关。
现在静脉注射免疫球蛋白(IVIg)和血浆置换已被证实可有效治疗GBS。然而,尽管有这些治疗,但许多患者仍经历了危重的病程、疼痛,残留有缺损。在本篇综述中,我们将总结GBS的免疫病理机制和临床特征,描述GBS现行的诊断标准,讨论脑脊液检查和神经传导检查的诊断价值,治疗的选择和预后(包括GBS患者新型的预测模型)。
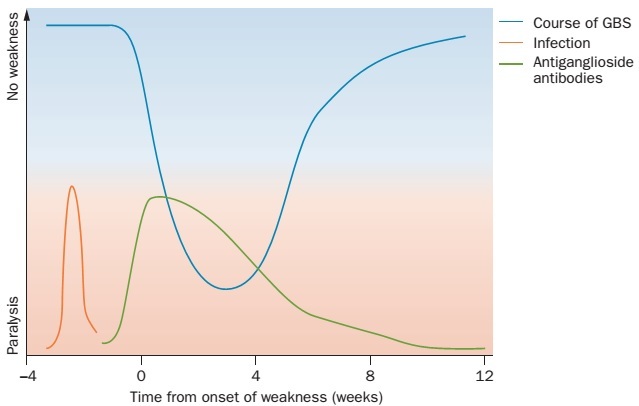
图1 . GBS的病程进展(蓝色)。多数GBS患者在肢体无力出现之前报告有前驱感染(红色);常可测及抗神经节苷脂抗体(绿色),但其水平随时间而降低;渐进性肢体无力在4周内达到高峰(常在2周内),恢复期可持续数周、数月甚至数年。Paralysis:肢体瘫痪无力。
一、流行病学
GBS是一种罕见病,发病率为0.81~1.89 (中位数为 1.11)/10万人/年,男性多见于女性(比率3:2)。全球的GBS发病率各异,譬如,巴西的发病率较低,为0.40/10万人/年;而库拉索岛和孟加拉国的发病率较高,为2.5/10万人/年。
GBS的儿童发病率(0.34~1.34/10万人/年)低于成人,发病率随着年龄增加而增高。尽管在库拉索岛、孟加拉国和中国的研究中发病率存在季节性波动,但多数研究中GBS的发病率在时间上较为稳定。
GBS患者AIDP和 AMAN的比率在全球范围内差异较大:在北美和欧洲,AIDP 是主要的亚型(60%~80%的患者);相反,在英国和西班牙,AIDP 占6%~7%;在亚洲、中美和南美为30%~65%。地理上的多样性可能导致了对某些感染的暴露不同;此外,由于不同地区个体和人群的基因多样性差异,基因易感性也各异。
上述差异不仅与特殊类型的GBS亚型相关,而且与疾病的病程和严重程度有关;需要进行大量人群的遗传学研究来探讨这些关系。
二、发病机制
GBS为感染后病变,或者说感染是GBS的前驱事件。2/3的患者报告在GBS发病前有呼吸系统或胃肠道感染的症状。近半数的GBS患者可发现存有某种特异性的前驱感染,而1/3的感染由空肠弯曲杆菌引起。其它可引起GBS相关前驱感染的病原体有:巨细胞病毒、EB病毒、肺炎支原体、流感嗜血杆菌和A型流感病毒。
约有2/3的GBS患者在该病发作前6周内有感染症状。这些感染可能会触发导致GBS的免疫反应。已有研究显示目前有6种病原体与GBS相关:空肠弯曲菌、巨细胞病毒、戊型肝炎病毒、肺炎支原体、EB病毒和寨卡病毒。另有研究认为,其他病原体也与GBS相关,但尚不清楚它们在GBS发病机理中的作用。但不存在前驱疾病并不能排除GBS的诊断,因为推定的感染或其他免疫刺激可能为亚临床性。
1976年首次报道疫苗与GBS相关,当时在接受“猪流感”疫苗的美国非军事人员中发现GBS风险增加了7.3倍。此后多种疫苗报道与GBS存在关联,但只有两项进一步的研究表明GBS与流感疫苗之间存在联系。这些研究表明,每增加一百万次接种,GBS病例将增加约一例。
免疫生物制剂(如肿瘤坏死因子拮抗剂、免疫检查点抑制剂或I型干扰素)与GBS已有相关性。其他事件,包括手术和恶性肿瘤,在时间上与GBS有关,但缺乏明确的生物学及流行病学证据。
值得注意的是,一项荷兰的病例对照研究显示,5%的GBS患者存在戊型肝炎病毒的前驱感染,而健康对照组中这一比率为0.5%。类似的,孟加拉国有10%的GBS患者存在戊型肝炎病毒的前驱感染,提示戊型肝炎病毒是GBS全球性的触发因素。
然而,尽管特异性急性感染和GBS之间存在强烈的关联,但感染后出现GBS的整体风险较低。每1000~5000的空肠弯曲杆菌感染患者中仅有1例在后继2个月内会发展为GBS。这解释了为什么说GBS是一种自发性疾病,尽管偶见空肠弯曲杆菌感染后GBS爆发的病例报道。
GBS发生机制的重要环节为空肠弯曲杆菌感染后,产生抗体与特异性神经节苷脂交互反应(图2),而这一抗体在非复杂性空肠弯曲杆菌性胃肠炎时并不产生。但交互反应性抗体只在易感个体中产生。
据报道,GBS患者中抗体可与多种神经节苷脂产生交互反应;但仅有空肠弯曲杆菌的一种亚型可含有脂寡糖,脂寡糖可模仿人类周围神经中神经节苷脂的糖肽类部分(图2)。
这种类似神经节苷脂的糖肽类结构的合成,依赖于一系列多态基因和酶;而这些多态基因和酶因空肠弯曲杆菌菌株的不同而异。空肠弯曲杆菌cstII基因的Thr51变异与GBS的发生有关,而Asn51变异与MFS有关。
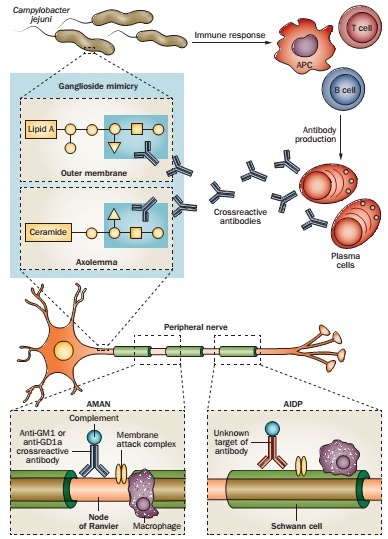
图2. GBS的免疫病理机制:分子模拟和抗神经节苷脂抗体。病原体(如空肠弯曲杆菌)的感染,可触发体液免疫和自发免疫应答,引起神经功能障碍和GBS症状。空肠弯曲杆菌外膜上的脂寡糖可诱发产生抗体(如周围神经上的抗GM1和抗GD1a抗体),抗体与神经节苷脂交互作用。引起AMAN的抗原位于Ranvier结上或附近。
抗GM1 和抗GD1a抗体与Ranvier结的轴膜结合,导致补体激活,继而膜攻击复合物形成、电压门控钠离子通道消失,该损害引起Ranvier结旁髓鞘脱离,神经传导障碍;继而巨噬细胞清除受损的轴索。引起AIDP的抗原位于髓鞘,抗体激活补体后导致Schwann细胞外表面上膜攻击复合物的形成,引起空泡样变性,巨噬细胞吞噬髓鞘。
一些抗体与特异性的GBS亚型及神经功能缺损有关,反映出人类周围神经中不同神经节苷脂的分布(表1)。空肠弯曲杆菌感染主要与GBS-AMAN或纯运动亚型有关,但并非只与这两个亚型有关。
AMAN患者常有血清抗GM1a、GM1b、GD1a 和GalNAc-GD1a神经节苷脂抗体。MFS患者或MFS-GBS重叠综合征的患者常有抗GD1b、GD3、GT1a 和 GQ1b神经节苷脂抗体,这与共济失调和眼肌麻痹有关。
表1 GBS的亚型、临床表现和相关抗体
|
GBS亚型 |
主要临床表现 |
NCS结果 |
抗体1 |
|
AIDP |
感觉运动型GBS,常有颅神经功能缺损伴自主功能障碍 |
脱髓鞘性多发性神经病 |
多种2 |
|
AMAN |
纯运动型GBS,颅神经受累罕见 |
轴索多发性神经病,感觉动作电位正常 |
GM1a, GM1b, GD1a, GalNAc-GD1a |
|
AMSAN |
类似重度的AMAN,但累及感觉纤维,引起感觉缺损 |
轴索多发性神经病,感觉动作电位降低或缺如 |
GM1,GD1a |
|
咽-颈-臂变异型 |
主要为口咽、面部、颈部和肩部肌肉无力 |
多数患者正常,有时可见上肢异常,多数为轴索型 |
GT1a>GQ1b>>GD1a |
|
MFS |
共济失调,眼肌麻痹,反射消失 |
多数患者正常,感觉传导分离性改变,可能出现H反射 |
GQ1b,GT1a |
注释:1:抗体主要为IgG,也可为IgM 和 IgA:;2:抗体在发病机制中的作用未明。
一项荷兰的研究显示,与巨细胞病毒感染有关的AIDP患者,其中20%有抗GM2抗体,尽管此类抗体也见于非复杂性巨细胞病毒感染的患者中。有趣的是,患者不仅有抗单个神经节苷脂的抗体,还有抗神经节苷脂复合物联合表位的抗体;该复合物位于细胞膜特殊的微区(脂筏)。
在空肠弯曲杆菌前驱感染的诱导下,针对神经节苷脂复合物的抗体还可与空肠弯曲杆菌的脂寡糖交互反应。抗糖脂复合体的抗体也见于AIDP的患者,但这些抗体在发病机制中的作用尚未明确。
与抗神经节苷脂抗体的出现相协同,补体的激活也参与了GBS神经退行性病变,该现象见于AMAN小鼠模型的朗飞氏节和运动神经末梢。钠离子通道簇、Ranvier结旁的轴突接头、结性细胞骨架、Schwann细胞微绒毛以及其它所有稳定钠离子通道簇的结构,在GBS疾病模型中均被补体激活所破坏。
GBS的小鼠模型还证明,阻止补体的激活可防止抗神经节苷脂介导的神经病变临床体征的出现。
空肠弯曲杆菌感染后发生的GBS,也可能依赖于患者相关的因素,这些因素可影响交互反应性抗体产生的易感性。GBS有5%的缓解率(明显高于机会性缓解),可支持该假设。病原体-宿主的相互作用是发生GBS的关键。
空肠弯曲杆菌的脂寡糖可与唾液酸结合性免疫球蛋白样凝集素-7(sialic acid-binding immunoglobulin-like lectin 7,siglec-7)结合,通过Toll受体4和CD14激活树突细胞。这些树突细胞可产生1型干扰素和肿瘤坏死因子,引起B细胞的增殖。该免疫反应可被基因多态性影响,但到目前为止,仅有小样本患者的研究关注了遗传因素。
有趣的是,meta分析发现,GBS和特异性的肿瘤坏死因子多态性之间具有中等程度的关联。此外,MBL2基因(编码甘露糖结合蛋白C)多态性和GBS的严重程度、临床结局之间的关联已被证实。未来需要大型队列的全基因组关联分析,研究宿主因素在GBS发病机制中的作用。
三、疫苗的作用
一些患者可在接受疫苗注射后短期内发生GBS,尽管此种情况较为罕见,但这引起了公众的关注。在1976年美国A型流感(H1N1)疫苗注射运动中,据估计疫苗相关的GBS风险为1/10万人;2009年的A型流感(H1N1)疫苗注射运动也存在相似的风险。
但全国和国际间的研究发现,疫苗相关的GBS风险较小:1.6个病例/100万疫苗接种者,这一频率在所有的季节性流感疫苗中相似。
实际上,疫苗可能会减轻获得GBS的风险,因为GBS常由流感等感染而诱发。据估计,流感感染后发生GBS的风险比接种流感疫苗后发生GBS的风险高出4~7倍。据观察,GBS的患者在接种流感疫苗后,无GBS的缓解。
由于这些原因,荷兰在现行的GBS患者疫苗接种指南中指出:GBS并非为流感疫苗接种的适应症;对于3个月前罹患GBS的患者来说,疫苗是安全的;疫苗接种后短期内不会发生GBS。
四、诊断
1. 何时怀疑GBS
(1). 典型临床表现
当出现双腿或双臂快速进展性无力而无明显中枢神经系统受累的表现或其他明显原因时,需考虑GBS。GBS典型的感觉运动表现为远端感觉异常或感觉丧失,伴有或继发腿部开始并发展到手臂和头颅肌肉的无力。大多数患者在就诊时或疾病高峰时腱反射降低或消失。自主神经异常很常见,包括血压或心率不稳、瞳孔异常以及肠/膀胱功能障碍。疼痛也很常见,可为肌肉、神经根性或神经性痛。疾病发作常为急性或亚急性,患者通常在2周内达到最大功能障碍。在发病后24h内或4周后达到最大功能障碍的患者,应考虑其他诊断。GBS一般为单相病程,尽管少数患者发生治疗相关波动和复发。
(2). 非典型临床表现
GBS也可以非典型方式出现。无力和感觉体征尽管总是双侧性,但可能为非对称性,或者以近端/远端为主,并且可从腿、手臂或同时在所有四肢开始。此外,严重和弥漫性疼痛或孤立性颅神经功能障碍可先于无力发作。尤其是年幼(<6岁)的儿童可能表现出非特异性或非典型临床特征,如定位模糊的疼痛、难以负重、易怒、脑膜炎或步态不稳。如果未能将这些体征识别为GBS的早期表现,可能会延误诊断。在少数具有非典型GBS的患者中,尤其是仅具有运动体征(纯运动变异)和在电生理检查中为急性运动轴索性神经病(AMAN)亚型的患者,在整个病程中可能会观察到正常的反射或反射活跃。
(3). 变异类型
GBS的变异类型有:无感觉体征的无力(纯运动变异);肌无力仅限于颅神经(双侧性面神经麻痹伴感觉异常)、上肢(咽-颈-臂肌无力)或下肢(截瘫型变异);MillerFisher综合征(MFS)包括眼肌麻痹、反射消失和共济失调(图1和表1)。总的来说,GBS变异很少为“单纯性”,并且经常与经典综合症有部分重叠或显示其他变异形式的典型特征。

图1 格林-巴利综合征(GBS)不同临床变异的症状模式。症状可能为纯运动、纯感觉(罕见)或运动和感觉的组合。共济失调可见于Miller Fisher综合征患者,而Bickerstaff脑干脑炎患者可同时出现意识下降和共济失调。尽管双侧面神经麻痹伴感觉异常、单纯感觉变异和Miller Fisher综合征已包括在GBS疾病谱中,但它们不符合GBS的诊断标准。
除了上面列出的变异之外,GBS谱还包含纯感觉共济失调、Bickerstaff脑干脑炎(BBE)和纯感觉变异,因为它们与GBS具有相同的临床或病理生理学特征。但由于这些临床变异类型不符合GBS的诊断标准(表2),因此尚存争论。除了运动症状和体征以外,纯感觉变异还具有经典GBS感觉运动形式的临床特征。单纯的感觉性共济失调和MFS具有重叠的临床特征,BBE患者通常表现出类似于MFS的症状,并随后出现脑干功能障碍的体征,包括意识障碍和锥体束征。与MFS患者相似,感觉性共济失调或BBE患者血清中可能出现针对GQ1b或其他神经节苷脂的IgG抗体。然而,是否单纯的感觉型GBS、单纯的感觉共济失调和BBE是GBS的变异,还是MFS的不完整形式尚存争议,当怀疑这些变异时需仔细进行相关检查
2. 诊断标准
GBS诊断十步法

1978年,美国国家神经学疾病与中风研究院在调查GBS和1976年~1977年猪流感疫苗注射运动之间的关系时,对GBS作了病例定义。GBS的初始诊断性标准出版于1981年,1990年做了修订;尽管该标准原先是为研究目的而设,但仍被广泛的用于临床实践中;该标准由坚决支持诊断的临床特征和怀疑为GBS的临床特征两部分组成(表2)。
在缺乏足够敏感性和特异性生物标记物的情况下,GBS的诊断主要基于临床病史和检查,以及辅助检查(如CSF检查和电生理检查)的支持。美国国立神经系统疾病和中风研究所(NINDS)于1978年(1990年修订)(表2)和2011年的Brighton Collaboration制定的GBS诊断标准可供参考。NINDS标准更适合临床医生,因为它们展现了典型和非典型GBS的临床特征,而Brighton Collaboration标准可以帮助临床医生对典型GBS或MFS进行鉴别。当怀疑GBS时,还必须牢记各种鉴别诊断,某些症状应引起其他诊断的怀疑。接下来将详细介绍辅助检查在确认GBS诊断中的作用。
表2. GBS的诊断标准
|
GBS诊断所需的临床表现 |
|
渐进性上、下肢无力(有时仅以下肢无力起病) |
|
无力肢体的反射消失(或腱反射减退) |
|
AIDP |
|
额外症状: 进展期持续数天至4周 症状相对对称 轻度的感觉症状或体征 累及颅神经,尤其是双侧面肌无力 自主神经功能障碍 疼痛(常见) |
|
NCS结果: 脱髓鞘特征(远端CMAP幅度>10% LLN) 远端运动潜伏期延长 运动神经传导速度下降 F波潜伏期、传导阻滞和时间弥散度增加 |
|
AMAN |
|
额外症状: 进展期持续数天至4周 症状相对对称 无感觉症状或体征 累及颅神经(罕见) 自主神经功能障碍 疼痛(偶见) |
|
NCS结果: 无脱髓鞘特征(或如果远端CMAP幅度< 10% LLN,仅1条神经出现脱髓鞘表现) 至少2条神经远端CMAP幅度< 80% LLN 可能出现一过性运动神经传导阻滞(由抗神经节苷脂抗体所致) |
|
不支持诊断为GBS的体征 |
|
脑脊液中单核细胞增多(>50 个/ μl)或出现多形核细胞 |
|
起病时严重的肺功能障碍,肢体无力 |
|
起病时严重的感觉体征,肢体无力 |
|
起病时膀胱或直肠功能障碍 |
|
起病时发热 |
|
脊髓感觉水平改变 |
|
缓慢进展的肢体无力、但无累及呼吸功能(考虑为亚急性炎症性脱髓鞘性多发性神经病或急性发作的慢性炎症性脱髓鞘性多神经病) |
|
持续性、不对称性的肢体无力 |
|
持续的膀胱或直肠功能障碍 |
注释:诊断GBS时不需要分类为AIDP或AMAN;尚不清楚AIDP和AMAN是否需要不同的治疗;定义脱髓鞘所需的传导减慢的具体标准在不同的分类系统中各异;LLN: lower limit of normal,正常值下限。
2011年,Brighton协作计划在研究GBS 和 2009~2010年H1N1猪流感疫苗接种运动的相关性时,对GBS做了新的病例定义;该标准从回顾性流行病学和疫苗安全性研究中发展而来。特异性较高而敏感性较差。Brighton标准将GBS定义为四个诊断确定性水平:第一级水平最高,第四级水平最低。Brighton标准区分GBS疑似患者,高度依赖于诊断性数据的完整程度。
一项荷兰的研究纳入了335名GBS患者,收集了全套的诊断性数据(根据Brighton标准,为第一级确定性水平所必需),将61%的患者为第一级水平,33%的患者为第二级水平,无第三级水平,6%的患者为第四级水平。
然而Brighton标准并非为临床诊断而设立。未来的研究需要进一步修订急性期GBS及其罕见变异的诊断标准。在临床实践中,1990版的标准为全球临床医师诊断GBS提供了良好的基础。
2. GBS的临床症状和亚型
GBS以快速进展的、对称性四肢无力伴反射减退或反射消失为特征。然而,GBS的起病、颅神经功能缺损的分布和程度、感觉症状、肌无力、共济失调、疼痛、自主神经功能障碍和病程特征高度各异。许多患者会有感觉缺损,如麻木和/或感觉减退。半数患者存在颅神经功能缺损,尤其是双侧面肌无力、吞咽困难或眼外肌运动障碍。
有较高比例的GBS患者(54%~89%)会出现疼痛,包括痛性感觉异常、背痛、肌肉痛和假性脑膜炎;在1/3的GBS病例中,这些症状可先于肌无力而出现。近25%的患者出现呼吸功能不全需要人工通气。自主神经功能异常(主要为心血管系统的调控障碍)可见于2/3的患者,但严重程度高度各异。约有1/3患者的步行功能轻度受损,但在病程中仍可行走。
GBS是一种单相病变,病程常在4周之内达到高峰。一项研究显示,80%的GBS患者在出现肌无力后的2周内,病情达到顶峰;97%的患者在4周内达到高峰。病情的进展期之后为平台期,在患者开始康复之前,平台期可持续2天~6个月(平均为7天)(图1)。
GBS不同的亚型,其临床表现、电生理和组织病理不同(表1和表2),目前了解的最多的两个亚型为AIDP 和 AMAN。
AIDP 为感觉运动型GBS,常伴有颅神经功能缺损、自主功能障碍和疼痛;电生理检查中以脱髓鞘性多发性神经病为特征。
相反,AMAN是纯运动型GBS,临床上和电生理检查中表现为轴索多发性神经病但无感觉缺损(约10%的AMAN患者有感觉症状)。AMAN的颅神经功能缺损较AIDP少见,但有自主神经功能障碍和疼痛。通常,AMAN的病程进展较AIDP更为迅速,由于轴索退行性病变而恢复期更长;但一些AMAN患者可从重度肢体无力中迅速恢复。AMAN常与空肠弯曲杆菌的先驱感染相关。
一些轴索性GBS的患者,感觉和运动纤维均受累及;此亚型称为运动感觉轴索性神经病(acute motor-sensory axonal neuropathy,AMSAN),可看做是AMAN的重度变异型。
MFS为GBS的少见亚型,以眼肌麻痹、共济失调和反射消失临床三联征为特征,多数患者可出现复视。通常,MFS患者的临床结局良好,但有些患者可发展有四肢无力和呼吸功能不全(称为MFS–GBS重叠综合征)。
肌电图远端肌肉复合动作电位(CMAP)检查显示,一些MFS患者存在亚临床肢体运动神经功能障碍。其它GBS的局灶性变异类型,如咽-颈-臂变异型,亦有报道。
3. 非典型GBS
约8%的GBS患者出现下肢轻瘫,令诊断变得更为复杂,这些患者在随访时仍有70%存留有下肢轻瘫;然而GBS患者罕见有确定性的不对性四肢无力。
尽管1990年的GBS诊断标准要求有反射减退或反射消失,但在一项队列研究中,求诊时9%的GBS患者无力上肢存在正常的腱反射,2%的的患者无力下肢腱反射正常;随访时,所有患者均表现出下肢反射减退或消失,但一些患者上肢的反射正常。一些GBS患者,尤其是AMAN亚型,腱反射可被完全保留甚至亢进,原因未明。
4. 其它检查
腰椎穿刺
疑似GBS的患者常行腰椎穿刺检查,当需要排除其它诊断而非确诊GBS时,此检查尤为重要。脑脊液中蛋白水平增高而细胞计数正常(称为蛋白-细胞分离),是GBS的特征性标志。
脑脊液检查主要用于排除GBS以外的其他无力原因,应在对患者进行初次评估时进行。GBS的经典表现是CSF蛋白水平升高和CSF细胞计数正常(称为蛋白-细胞分离)。然而,在发病后第1周,30%-50%的患者CSF蛋白水平可正常,而在第2周10%-30%的患者可正常。因此,CSF蛋白水平正常并不排除GBS。明显的细胞增多(> 50个细胞/μl)提示存在其他病变,如软脑膜恶性肿瘤或脊髓、神经根的感染性或炎性疾病。轻度细胞增多症(10-50个细胞/μl)尽管可见于GBS,但仍应促使临床医生考虑其他诊断,如多发性神经根炎的感染原因。
但必须有蛋白-细胞才能诊断为GBS,这一观念是错误的,仅有64%的GBS患者可见该现象。约有50% 的患者,在四肢无力发病后的头3天可见脑脊液蛋白水平升高,发病第一周后在80%的患者中可见蛋白水平增高。
当脑脊液细胞计数>50个/ μl时,可排除GBS的可能性,考虑为软脑膜恶性肿瘤、淋巴瘤、巨细胞病毒脊髓神经根炎、HIV多发性神经病和脊髓灰质炎等其它诊断。倘若脑脊液中的蛋白水平正常,由于蛋白-细胞分离并非为诊断GBS所必须,不推荐做重复腰椎穿刺。
此外,可能由于渗漏或无菌性脑膜炎,高剂量IVIG治疗可增加脑脊液中的蛋白水平和细胞计数,此时重复性腰椎穿刺的结果可能会干扰诊断。
肌肉和神经电生理
诊断GBS不需要电生理检查。但建议尽可能进行这些检查,因为它们有助于支持诊断,尤其是对于非典型表现的患者。通常GBS患者的电生理检查会发现感觉运动性多神经根神经病或多发性神经病,表现为传导速度降低、感觉和运动诱发幅度降低、时间分散异常和/或部分运动传导阻滞。GBS的典型特征是“腓肠神经豁免”,其中腓肠感觉神经动作电位正常,而正中神经和尺神经感觉神经动作电位异常甚至消失。但在疾病早期(症状发作1周内)或最初表现为近端无力、病情较轻、进展缓慢或临床变异的患者中,电生理检查结果可为正常。在这些患者中,2-3周后复查电生理可能会有所帮助。对于MFS患者,电生理检查结果通常为正常,或仅显示感觉神经动作电位幅度降低。此外,电生理检查还可区分经典GBS的三种亚型:急性炎症性脱髓鞘性多发性神经病(AIDP)、急性运动轴索性神经病(AMAN)和急性运动感觉轴索性神经病(AMSAN)。
神经传导检查(Nerve conduction studies,NCS)可辅助GBS的临床诊断,鉴别轴索性和脱髓鞘性亚型。在疾病的早期,有时诊断GBS较为困难,尤其是仍存有反射或肢体无力的分布不符合经典模式(譬如出现下肢轻瘫)时。NCS在亚临床病变中可显示出局部(如上肢)的异常,NCS发现的周围神经病或多发性神经根病有助于GBS的确诊。
肢体无力发病后2周,神经传导的异常可达到高峰。然而,如需NCS确诊GBS的疑似病例,则2周的时间太长。尽管在病程早期可行NCS检查,但此时结果可能正常。通常,NCS异常最早表现为传导延迟或F波缺如,随着病程的进展还会出现其它异常。为提高NCS的诊断率,至少要做4条运动神经和3条感觉神经,还要检查F波。
NCS的异常发现因GBS亚型(AIDP、AMAN或 AMSAN)而异。AIDP的患者,NCS为脱髓鞘性表现,包括远端运动潜伏期延长、神经传导速度减慢、F波潜伏期延长、时间弥散度增加和传导阻滞,腓肠神经感觉电位常有保留。
轴索性GBS(AMAN 或AMSAN)的NCS表现包括运动和/感觉电位的振幅下降,无脱髓鞘性特征。感觉神经检查有助于鉴别AMAN和 AMSAN。AMAN的神经电生理结果可十分复杂,一些患者可出现一过性传导阻滞或传导减慢,随病程进展而快速恢复,该现象称为可逆性传导衰竭。
可逆性传导衰竭可模拟脱髓鞘性表现,这可能是由于抗神经节苷脂抗体损害了Ranvier结的传导所致。除了可逆性传导衰竭之外,还可在轴索性GBS急性期观察到其它提示为脱髓鞘病变的NCS表现。
有可逆性传导衰竭表现的患者可被误诊为AIDP而非AMAN,系列NCS检查有助于鉴别两者。譬如,在一项意大利的研究中,24%最初被诊断为AIDP的患者,在系列NCS检查后被归为AMAN型。
由于尚无神经电生理确诊GBS亚型的统一标准,因此存在多个分类系统。目前亚型的分类对治疗无影响,轴索性GBS和脱髓鞘性GBS的治疗,原则上相似。
影像学检查
MRI不是GBS常规诊断评估的一部分,但可能会有所帮助,特别是对于排除鉴别诊断。神经根增强是GBS一种非特异性但较敏感的特征,可以支持GBS的诊断,尤其是对于临床和电生理评估均存在困难的幼儿。另一种对GBS诊断有潜在价值的工具是对周围神经进行超声检查,有研究显示在疾病早期,颈神经根会增大,提示脊髓根部炎症作为早期病理机制的重要性。因此,尽管需要进一步的验证,但该技术可能有助于在疾病早期对GBS进行诊断。
检测抗神经节苷脂抗体
尽管GBS的发病机制中涉及抗神经节苷脂抗体,但其在诊断中的作用还未确定。一般而言,特异性抗体的滴度较低,意味着检测的阴性预测值也较低,因此阴性结果并不能排除GBS的可能性。此外,检测的阳性预测值的意义有限,因为其它疾病也会出现抗神经节苷脂抗体。
但抗GQ1b抗体,至少90%的MFS患者可出现该抗体;抗GM1和 抗GD1a IgG抗体也常见于AMAN患者;这些抗体的检测有助于诊断。抗神经节苷脂抗体的检测方法正在迅速发展,在不久的未来会出现敏感性和特异性更高的检测方法。
5. 鉴别诊断
具有GBS典型特性的患者,诊断较为容易;但对于非典型表现的患者来说,有时难以诊断。具有典型表现的患者,也应行腰椎穿刺检查排除其它诊断。GBS的鉴别诊断包括感染性疾病、恶性肿瘤和神经肌肉接头病变。
对于脑脊液细胞计数增高的患者,应考虑巨细胞病毒或HIV引起的脊髓神经根炎症、横贯性脊髓炎、Lyme病、软脑膜恶性肿瘤或脊髓灰质炎等鉴别诊断。实验室检查也有助于查明GBS样症状的病变,如电解质紊乱(低钾血症)和维生素B1缺乏。
纯运动症状的患者,鉴别诊断应该考虑重症肌无力、多发性肌炎和皮肌炎、脊髓灰质炎、高镁血症、卟啉症、肉毒中毒、铅中毒或有机磷重度。NCS有助于鉴别多发性神经病、肌病、前角细胞病变(脊髓灰质炎)和神经肌肉接头疾病。
当对下肢轻瘫或脊髓感觉水平异常的患者考虑为GBS诊断时,应行脊髓MRI和脑脊液检查排除脊髓卡压或横贯性脊髓炎。NCS有助于确诊,脱髓鞘性多发性神经病或临床检查正常的上肢出现神经传导异常,都提示为GBS。MRI对神经根的检查可支持GBS诊断。对存在膀胱或直肠功能障碍的患者,鉴别诊断包括脊髓或马尾卡压、横贯性脊髓炎。
对于不对性肢体无力的患者,鉴别诊断应考虑为血管炎性神经病、多发性单神经病、Lyme病、白喉、脊髓灰质炎和软脑膜恶性肿瘤。倘若呼吸衰竭与肢体无力的程度不相称,则可哦哎出重症肌无力、高镁血症、低磷血症、高位颈髓髓内病变、脊髓灰质炎和肉毒中毒。
6. 儿童GBS的诊断
儿童和成人GBS的临床表现和结局有所不同,诊断儿童GBS(尤其是<6岁的儿童)较为困难。儿童出现疼痛、步行困难或拒绝步行时,应怀疑为GBS。然而,仅1/3的学龄前儿童GBS患者得到了正确的诊断。儿童GBS患者最初常被诊断为脑膜炎、髋关节炎或病毒感染所致的身体不适。
此外,儿童GBS的诊断常有延迟;对于年龄<6岁的学龄前儿童,诊断的延迟常在2周以上;倘若此时对这些儿童的监控不足,则将导致紧急插管甚至死亡。
五、评价量表
在过去的数十年,多数的临床试验采用GBS残疾量表评价临床结局。GBS残疾量表分为6个水平:0(健康);1(轻微症状,可以跑);2(无辅助下可步行10米,但不能跑);3(可在辅助下步行10米,穿过空旷区域);4(卧床或依赖轮椅);5(需要辅助通气);6(死亡)。
GBS的评估量表必须具有效度、敏感度和信度,可以捕捉到病程中的亚临床改变。周围神经病结局测量小组(PERINOMS)调查了大量的评估量表,在欧洲神经肌肉中心会议上,PERINOMS通过了一项合意,推荐使用GBS残疾量表和Rasch-built 整体残疾量表测量残疾程度,使用Medical Research Council总分(0~60)和新Rasch-built Medical Research Council分数来测量肌肉力量。
六、治疗
GBS的治疗常联合多学科的医疗照顾和免疫治疗(图3)。GBS已证实的有效治疗为IVIg和血浆置换。倘若患者无辅助下未能步行10米(GBS残疾量表≥3级),则可启动免疫疗法。血浆置换和IVIg具有多效免疫调节作用,但尚不清楚是何种作用对GBS的治疗起效,也不清楚所有患者和所有GBS亚型是否存在相同的治疗机制。
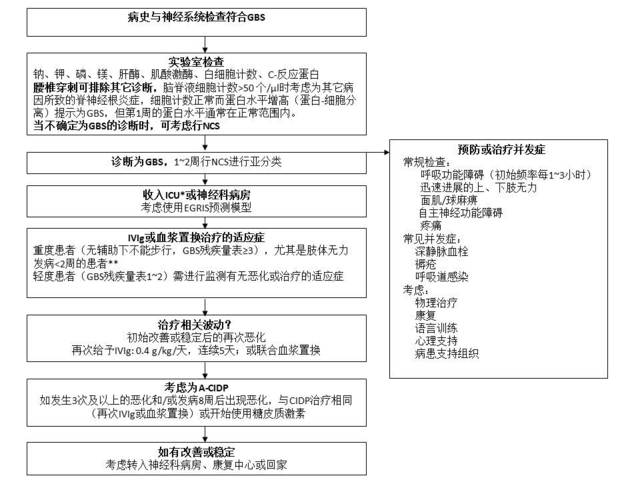
图3. GBS的诊断和治疗。
*:进入ICU的适应症为快速进展性肢体无力伴有呼吸功能受损需要机械通气(肺活量<20 ml/kg)、吞咽困难(重度肌无力)和严重的自主神经功能障碍。
**:医师决定是否存在治疗的适应症,譬如快速进展的肢体无力、轻度肢体无力伴严重的自主神经功能障碍或重度吞咽障碍;IVIg,发病2周内开始治疗,0.4 g/kg /天,5天;血浆置换,5次,2周内完成,置换总剂量为5倍的血浆量。
IVIg治疗可抑制Fc介导的免疫细胞的激活,可将抗神经节苷脂抗体与其神经靶点、或与局部激活的补体相结合。GBS患者血清IgG Fc糖基化与疾病严重程度相关,可影响IVIg的免疫调节作用。
血浆置换可清除神经毒性抗体、补体因子和其它体液中的炎性介质。对于不能无辅助下步行的患者(GBS残疾量表≥3级)来说,在肢体无力发病后的前4周之内,血浆置换可起效;但在初始2周内的效果最佳。常用的血浆置换方案为2周内给予5次治疗;对轻度残疾的患者(可步行),2次血浆置换后,运动功能就可迅速恢复。
IVIg对不能无辅助下步行的患者(GBS残疾量表≥3级)以及发病2周内的患者有效。随机对照研究显示,0.4 g/kg/天、连续5天或1g/kg/天、连续2天的IVIg治疗十分有效,但尚未评估前一种方案是否优于后者。但一项研究显示,与接受5天方案的相比,接受2天方案的儿童发生治疗相关波动的频率更高。
IVIg(每天0.4μg/kg体重,连续5天)和血浆交换(200-250ml血浆/ kg体重,分五个疗程)对GBS均有效,且疗效相当。IVIg和血浆置换的不良事件风险相当,尽管早期研究表明血浆置换比IVIg中止的可能性更大。由于与血浆置换相比,IVIg的给药更容易,并且使用范围更广,因此通常是首选治疗方法。除了IVIg和血浆置换,没有其他方法或药物被证明可有效治疗GBS。尽管预计皮质类固醇对减少炎症和GBS的疾病进展有益,但八项关于皮质类固醇对GBS疗效的随机对照试验未显示出明显的益处,口服皮质类固醇治疗甚至显示出负作用。此外,血浆置换后给予IVIg并没有比单独使用任何一种治疗更有效。在IVIg治疗的患者中,尚无足够的证据证明静脉注射甲基泼尼松龙的附加治疗效果。在资源有限的临床环境中,小体积血浆置换可能是传统血浆置换的一种经济且相对安全的替代方法,但是在进一步的试验中确定其有效性之前,不建议将该方法普遍使用。患有持续感染的GBS患者可以考虑采用抗微生物或抗病毒治疗;但先前的感染通常在无力发作之前就已缓解。
治疗的选择依赖于患者因素和社会经济因素。譬如,血浆置换需要特殊的设备,且不是所有的医院都可做血浆置换。此外,血浆置换在儿童患者中难以实施;由于大血容量的置换,心血管自主神经功能紊乱的患者必须加强护理。
IVIg的治疗费用要比血浆置换高出2倍,因此低收入国家较少采用;而大多数的临床试验也是在西欧和北美开展的,这些临床试验的结果可能不适宜于其它地区,尤其是AMAN的高发地区。
AMAN患者是否应该结合与AIDP相同的治疗方法,仍未明确。一项小型研究提示,AMAN患者采用血浆置换治疗的临床结局好于IVIg治疗,且血浆置换疗法最具成本-效益,但该研究存在方法学上的缺陷。在一些国家和地区,倘若缺少血浆置换设备或患者无法负担费用,则小剂量的血浆置换或置换输血是成本较低的治疗方法,但目前还缺少此类治疗的对照研究。
血浆置换后联合IVIg治疗的效果并未显著强于单用血浆置换或IVIg治疗。口服类固醇或静脉内应用甲强龙不能令GBS患者获益。IVIg联合甲强龙治疗的效果并不比单用IVIg更佳,尽管该联合治疗可能有些短期效果。一项小型的随机安慰剂对照研究显示IFN-β1和脑源性神经营养因子无效,另一项研究将霉酚酸酯联合标准IVIg治疗6周与单用IVIg比较,也显示并无增益。
尚无针对MFS患者的随机对照试验。一项回顾性分析显示,接受IVIg治疗的MFS患者,较血浆置换或非免疫治疗的患者,开始恢复的时间稍早;但三组最终的结局相同;实际上,无论接受何种治疗,所有的MFS患者都完全恢复。
由于缺乏MFS患者IVIg治疗或血浆置换有效性的证据,以及患者可自然恢复,目前MFS患者可不采用免疫疗法。然而,MFS-GBS重叠综合征的患者病情较重,IVIg或血浆置换仍为治疗的选择。需要注意的是,尚无大型随机对照研究明确儿童GBS患者的最佳治疗策略,因此对儿童患者常给予与成人GBS相同的治疗。
何时进入ICU?
需要进入重症监护病房(ICU)的原因包括:呼吸窘迫加重,即将出现呼吸功能不全(定义为呼吸窘迫的临床体征,包括休息或谈话时的呼吸困难、单次呼吸无法数到15、使用辅助呼吸肌、呼吸或心率增加、肺活量<15– 20ml / kg或<1L、动脉血气或脉搏血氧饱和度异常);严重的自主性心血管功能异常(如心律不齐或血压明显变化);严重的吞咽功能障碍或咳嗽反射减弱;以及肌无力迅速发展。
由于入院第1周内有22%的GBS患者需要机械通气,因此必须尽早确定存在呼吸衰竭风险的患者。为此,可使用Erasmus GBS呼吸功能不全评分(EGRIS)量表,进行评估1周内需要通气的可能性(1–90%)(表3)。机械通气时间延长的危险因素包括:插管后1周无法将手臂从床上抬起,以及电生理检查可见轴突亚型或难于刺激的神经。有这些危险因素的患者应考虑早期气管切开术。
何时开始治疗?
如果患者无法独立行走10m,则应开始免疫调节治疗。对于仍可独立行走的患者,证据有限,但应考虑治疗,特别是如果这些患者出现快速进展的无力或其他严重症状,如植物神经功能障碍、延髓衰竭或呼吸功能不全。临床试验表明,在无力发作后2周内开始静脉注射免疫球蛋白(IVIg)和在4周内开始进行血浆置换可使患者获益。在这些时间段之外,缺乏有效性的证据。
特殊人群的治疗
(1)GBS变异
单纯MFS患者往往病程相对较轻,大多数患者在6个月内无需治疗即可完全康复。因此,通常不建议对患者给予治疗,但应密切监测患者,因为其可能会出现肢体无力、延髓或面神经麻痹或呼吸衰竭。BBE病程的严重性证明可使用IVIg或血浆置换,尽管证据有限。对于其他临床变异类型,目前尚无相关报道,但许多专家建议IVIg或血浆置换。
(2)孕妇
在怀孕期间,IVIg和血浆置换均非禁忌。但由于血浆置换需要更多的考虑和监测,因此IVIg可能是首选。
(3)儿童
儿童的治疗跟成人差不多。有关儿童血浆置换和IVIg的证据有限。但是,由于血浆置换只在有使用经验的中心提供,并且似乎比IVIg更可能产生不适感和并发症,因此IVIg通常是儿童GBS的一线治疗。尽管一些儿科中心建议2g/kg(体重)的IVIg 2天方案,而不是5天成人标准给药方案,但一项研究表明2天方案的治疗相关波动发生率更高。
GBS重要的支持性照顾
GBS进展期的患者需要住院治疗,治疗的重点在于监测病程的进展以及避免肢体无力、制动、呼吸功能不全、自主神经功能障碍和疼痛相关的并发症。GBS患者需要多学科的支持性照顾来预防或管理多样的并发症(图3)。
照顾的内容包括监测呼吸功能,譬如呼吸深度、呼吸频率和咳嗽的强度,但监测的频率尚无研究,可以根据病情的恶化速率每1~4小时监测一次;采用选择性消化道去污染可缩短通气的持续时间。
其它需要保持警惕的问题有预防深静脉血栓、心功能和血液动力学监测、疼痛的管理、膀胱和直肠功能障碍的管理、物理治疗、康复和心理支持。GBS患者及家属还可联系一些国际性的患者组织。
疾病进展的监测
对于GBS患者需要定期评估以监测疾病的进展和并发症的发生。首先,建议常规测量呼吸功能,因为并非所有呼吸功能不全的患者都会有呼吸困难的临床症状。这些呼吸功能的测量包括辅助呼吸肌的使用、一次呼吸的计数(一次呼吸计数≤19表示需要机械通气)、肺活量以及最大吸气和呼气压力。临床医生应考虑使用“20/30/40规则”,如果肺活量<20ml / kg、最大吸气压力<30cmH2O或最大呼气压力<40cmH2O,则认为患者有呼吸衰竭的风险。其次,应使用医学研究理事会分级量表或类似量表来评估颈部、手臂和腿部肌肉力量,并应根据GBS残疾量表评估功能障碍。第三,应监测患者的吞咽和咳嗽困难。最后,应该通过心电图和监测心率、血压以及肠/膀胱功能来评估自主神经功能障碍。
监测的性质和频率取决于病情恶化的速度、是否存在自主神经功能障碍、疾病的阶段以及医疗机构。应对每位患者进行仔细评估。GBS患者中多达2/3的死亡发生在恢复阶段,主要因心血管和呼吸功能障碍所致。因此,建议临床医生在此阶段保持警惕,并监测潜在的心律不齐、血压变化、呼吸窘迫。对于刚离开ICU的患者和有心血管危险因素的患者,这种监测尤为重要。
早期并发症的管理
GBS的并发症可导致严重的残疾和死亡。其中一些并发症包括压力性溃疡、医院获得性感染(如肺炎或泌尿道感染)和深静脉血栓形成。其他并发症是GBS所特有,如延髓性麻痹患者无法安全吞咽;面瘫患者的角膜溃疡;肢体无力患者的肢体挛缩、骨化和压力性麻痹。GBS患者也经常出现疼痛、幻觉、焦虑和抑郁,护理人员应特别询问患者是否存在这些症状。
初始改善后的恶化
约有10%的GBS患者在接受IVIg或血浆置换后,病情初始改善或稳定之后出现恶化,该现象称为治疗相关波动。再次进行IVIg或血浆置换后,患者通常得到改善;尽管尚无随机对照试验证再次治疗的效果,但这已成为常规的临床实践。
根据临床经验,我们推荐存在治疗相关波动的患者进行IVIg再次治疗(2 g/kg ,连续 5 天);此类患者可能由于延长的自发免疫反应而引起神经损害或功能障碍,需要延长治疗。
进展期延长的GBS
有些患者病程可多次恶化或进展期超过4周,则应怀疑此类患者是否真为GBS还是急性发作的慢性炎症性脱髓鞘性多神经病(A-CIDP)。初始诊断为GBS的患者,若出现3次及以上的临床恶化或发病后病情恶化超过8周,则应考虑为A-CIDP(表3)。
确定病情的恶化为迟发性或新发型非常重要,因此GBS治疗相关波动的患者再次IVIg治疗就可改善,而A-CIDP患者则需要采用IVIg慢性维持性治疗或换成糖皮质激素治疗。
表3. GBS、GBS-TRF、A-CIDP 和 CIDP的鉴别诊断
|
特征 |
GBS |
GBS-TRF |
A-CIDP |
CIDP |
|
达到峰值的时间 |
<2周(最大4周) |
<2周(最大4周) |
4~8周,随后进行性恶化 |
>8周 |
|
病程 |
单相型 |
8周内1~2次恶化 |
>2次恶化或8周后恶化 |
进行性、阶梯式或波动性 |
|
严重程度 |
患者之间高度异性,从轻度症状至瘫痪 |
患者之间高度异性,从轻度症状至瘫痪 |
多数为中度 |
多数为中度的、远端和近端肢体无力 |
|
依赖通气 |
20~30% |
20~30% |
几乎不需要 |
几乎不需要 |
|
颅神经缺损 |
常见 |
常见 |
偶见 |
偶见 |
|
对IVIg的反应 |
好 |
好,有波动 |
不同 |
好 |
|
EMG/NCS |
有时第一次EMG/NCS检查无法分类 |
有时第一次EMG/NCS检查无法分类 |
第一次EMG/NCS常为脱髓鞘性多发性神经病 |
脱髓鞘性 |
|
治疗 |
IVIg或血浆置换 |
重复IVIg或血浆置换 |
IVIg或血浆置换,确诊为CIDP则需泼尼松维持治疗 |
IVIg,泼尼松或血浆置换 |
注:GBS-TRF: Guillain–Barré syndrome with treatment-related fluctuation,格林巴利综合征伴治疗相关波动
七、残留的缺损
GBS成人患者常遗有功能上的缺损,严重影响了日常活动和生活质量。譬如,发病后6个月,约20%的GBS患者在无辅助下仍不能行走。功能的改善多出现在发病后第一年,在此期之后仍可有所改善。遗留的最常见的缺损为肌力下降、感觉症状、疲劳和疼痛,许多患者必须改变生活方式、工作和社交活动。
然而,即使患者具有较好的功能性结局(GBS残疾量表≤1),心理社会健康状态也可能降低。
一些证据显示,高强度、多学科的康复科建设GBS患者的残疾,改善生活质量。GBS儿童患者的结局可能较成人更佳,但儿童患者影响日常生活的长期残留缺损还未被详细研究。
多数GBS患者可有长期持续的疲劳。可用疲劳程度量表和Rasch-built疲劳量表评价疲劳,目前引起疲劳的病理生理机制还未知,但可能与轴索的丧失有关。一项非对照非双盲性研究显示,物理训练对神经功能恢复良好、但仍有重度疲劳的GBS患者有效。
GBS患者常有中至重度疼痛。一项前瞻性随访研究显示,2/3的患者报告在发病后的前3周有疼痛,1/3的患者在1年后仍有疼痛。高达89%的患者在病程中的某个时期有疼痛,约有1/3的患者疼痛的出现先于肢体无力。疼痛最常见的部位为四肢或背部,主要为肌肉痛、痛性感觉异常、神经根性痛、关节痛或假性脑膜炎。倘若整个病程中均有疼痛,则须提高警惕。
可用神经性镇痛剂治疗患者,但并非对所有患者均有效。一项针对GBS患者药物治疗疼痛的Cochrance评价显示,缺乏支持使用镇痛剂缓解疼痛的证据。引起GBS疼痛的原因可能是多样的,在不同的病程阶段也有不同。值得注意的是,GBS患者远端肢体的表皮神经纤维密度降低,这与疼痛的发生率以及疼痛的严重程度均有关(但与自主神经功能障碍无关)。
自主神经功能障碍主要见于GBS的急性期,但也可见于恢复期;这对GBS患者来说是一个严重的问题,可引起猝死。有报道,156名GBS患者中,出现心动过速(38%)、高血压(69%)、胃肠道功能障碍(45%)和膀胱功能障碍(19%)。严重心血管系统功能障碍的患者,其血压可迅速改变,出现心律失常,有时需要安装起搏器。
在儿童患者中,半数轻度病情的患者出现自主神经功能障碍,且与疾病的严重程度无关,提示自主神经功能障碍较为常见,可发生于任何程度的GBS中。
GBS患者的自主神经功能障碍反映了交感和/或副交感神经支配的功能障碍,但精确的免疫病理机制尚未被阐明,特别是特殊的抗神经节苷脂抗体和自主神经功能障碍之间的关系需要进一步研究。我们并没有观察到表皮神经纤维密度的减少与自主神经障碍障碍之间的相关关系。
一些患者出现严重的自主神经功能障碍(瞳孔对光反射消失、过度出汗),而另一些患者可能存在重度的心律失常,这其中的原因未明,且无法从临床的角度去预测。患者从ICU转出后,需要严密的监测自主功能障碍的表现。
八、预后及结局
GBS的临床病程和结局高度各异,对其的精确预测可令临床医师提供个体化的支持性照顾和治疗、以及告知患者及其家属未来临床病程的发展。
大多数GBS患者,即使是在疾病高峰出现四肢瘫痪或需要长时间机械通气,也可很大程度地康复,尤其是发病后的第1年。大约80%的GBS患者在发病后6个月恢复独立行走能力。可以使用改良的Erasmus GBS结局评分(mEGOS)计算个别患者恢复步行能力的可能性。
尽管GBS患者的总体预后较好,但仍有3%-10%的病例死亡,最常见的原因是心血管和呼吸系统并发症,在急性和恢复期均可发生。死亡的危险因素包括高龄和发病时的严重程度。长期残留的不适也很常见,可能包括神经性疼痛、无力和疲劳。GBS的复发很少见,见于2–5%的患者。尽管GBS并不是疫苗的严格禁忌症,但许多疫苗都带有关于GBS的警告。因此,在接种疫苗时需权衡利弊。
康复计划
GBS患者可能会遇到一系列长期的后遗症问题,包括运动和感觉功能恢复不全、疲劳、疼痛和心理疾病。
1. 机体功能
与康复专家、理疗师和职业治疗师安排康复计划是迈向康复的关键一步。康复计划应旨在康复的早期阶段减少残疾,随后将运动和感觉功能以及身体状况恢复到疾病前水平。GBS患者的运动计划包括全关节运动、固定自行车运动以及步行和力量训练。但是,必须监控运动强度,因为过度劳累会导致疲劳。
2. 疲劳
与残余运动功能障碍无关的疲劳见于60%-80%的GBS患者,并且通常是最致残的主述之一。在断定患者的疲劳是GBS的后遗症之前,应考虑其他原因。与身体机能的恢复一样,分级的有监督的锻炼计划已显示出对减轻疲劳的作用。
3. 疼痛
至少1/3的GBS患者在疾病发作后1年出现严重疼痛,并且可持续超过10年。GBS的慢性疼痛表现为下背部和四肢肌肉疼痛、痛性感觉异常、关节痛和神经根疼痛。管理策略包括鼓励活动和服用治疗神经性或伤害性疼痛的药物。
4. 心理问题
通常在先前健康的个体中,身体机能的迅速丧失可能引起焦虑和/或抑郁。早期识别和处理心理问题对GBS患者很重要。
通气需求的预测因子
三个大型的研究预测了GBS患者出现呼吸功能不全的可能性。一项法国的研究纳入了722名患者,结果发现发病到入院时间<7天、咳嗽不能、站立不能、从床面上举肘或举头不能、肝酶水平增高可能为需要人工通气的预测因子。另一项来自法国的研究发现,腓神经传导阻滞和肺活量低于呼吸衰竭的高风险相关。
荷兰研究的一项研究,通过对397名GBS患者进行队列分析,研制了Erasmus GBS呼吸功能不全分数(EGRIS)。EGRIS是一个精确预测的模型,可用于急诊环境中,在GBS患者入院后第一周内预测呼吸功能不全的可能性。
该模型含有以下参数:肢体无力的程度(用Medical Research Council总分评价)、肢体无力的发病时间与入院时间之间的天数、面肌无力和/球麻痹。倘若患者发生呼吸功能不全的预测机会较高,则建议入住重症监护室而非普通的神经科病房。
预测患者能成功撤离通气的因子有:年龄<60岁、无自主神经功能障碍、肺活量>20 ml/kg或改善4 ml/kg。自主神经功能障碍、高龄和肺部并存疾病都与机械通气时间延长以及需要气管插管相关。
长期结局较差的预测因子
多数研究将结局较差定义为6个月或12个月后GBS残疾量表≥3级;发病前腹泻、高龄等因素为临床结局差的预测因子。
Erasmus GBS结局分数(GBS Outcome Score,EGOS)可有效预测患者的长期结局,该模型基于三个临床参数:年龄、入院2周后的GBS残疾量表的分数和前驱腹泻。
但是EGOS需要在入院2周后才可进行,而这2周的延迟令医师无法根据预测的结局而调整患者的治疗。相反,改良EGOS可用于早期评估GBS,在入院时和入院1周后根据年龄、肢体无力的程度(用Medical Research Council总分评价)和前驱腹泻预测临床结局。
电生理结果
除了用于诊断,电生理的结果还与预后相关。在一项NCS研究中,有脱髓鞘改变的患者更需要机械通气。低CMAPs可预测较差的临床结局。但是这些研究显示轴索神经病变并非为预测较差临床结局所必需,因为AMAN患者可缓慢、不完全的恢复或迅速恢复(一过性传导阻滞的恢复所致)。
未来需要进一步研究确定传导阻滞、抗神经节苷脂抗体、治疗效果和临床结局之间的关系。
死亡率的预测
GBS的死亡率在3%~7%。死亡风险增高的预测因子为高龄、病情严重、心肺并发症、机械通气和全身感染。一项研究显示,多数死亡发生在发病的30天之后,另一项研究也显示多数患者死于恢复期。因此,GBS恢复期的患者以及从重症监护室内转出的患者仍需紧密的观察和支持性照顾。最常见的死因为呼吸功能不全、肺部感染、自主神经功能障碍和心脏骤停。
九、进展中的研究
在首次被描述后的近一个世纪以来,GBS仍为一种威胁生命性的疾病,至少20%的患者临床结局较差。从临床观点来看,急需下列改善:
发展适合临床实践、临床试验和疫苗安全性研究所应用的诊断标准;在全球范围内确定GBS带来的疾病负担;研制GBS结局的评价方法;探讨引起GBS的患者相关因素;研究临床病程和结局的生物学以及临床的预测因子;最重要的是发展最具效果和特异性的治疗方法以及支持性照顾的程序。
上述目标只有开展大型的国际间、多学科的合作研究才可完成,如2012年开展的国际GBS结局研究(IGOS)。IGOS在全球范围内至少招募1000个GBS患者,随访1~3年,收集组织样本和标准化的临床数据,目前已有17个国家(阿根廷、澳大利亚、孟加拉国、比利时、加拿大、丹麦、法国、德国、意大利、日本、马来西亚、荷兰、西班牙、南非、中国台湾、英国和美国)成立了研究中心。
此外,荷兰也在开展一项随机对照研究,SID-GBS。该临床试验调查给予第一疗程IVIg后预后较差的GBS患者(定义为接受标准5日IVIg疗法后的1周,改良EGOS分数6~12分),接受第二疗程的IVIg治疗效果。另一项相关的国际性研究,I-SID-GBS,也采用了相同的方法;I-SID-GBS为IGOS的一部分,由炎性神经病联盟所主导。
一项新随机安慰剂对照试验即将开展,研究依库丽单克隆抗体(eculizumab)对不能步行的早期GBS患者的疗效。
该研究根据GBS动物模型的研究结果,抗神经节苷脂抗体可对Ranvier结、神经末梢和突触旁Schwann细胞诱导产生补体依赖性损伤,但补体对神经末梢的恶性作用可被依库丽单抗阻滞。依库丽是人源性单克隆抗体,可与补体因子C5高亲和性结合,防止C5裂解成C5a和促炎性的C5b-9复合体。
目前还急需针对疼痛治疗、神经再生和改善GBS患者临床结局的研究。
十、小结
GBS是一种异质性、病情较重的疾病,尽管非特异性免疫治疗对多数患者均有效,但病程中仍须改善治疗和耐心的照顾,优化支持性的医疗照顾对预防或治疗疾病相关的并发症十分重要。新颖的预测模型的发展,满足了个体化治疗的需求。
尽管已知GBS不同亚型的免疫病理机制,但仍需要此方面的进一步研究。在过去数年,已开展了数个全球性大规模的临床试验,这将有助于进一步了解该综合征、优化患者的医疗照顾。
原始出处:
Leonhard SE, Mandarakas MR, Gondim FAA, Bateman K, Ferreira MLB, Cornblath DR, van DoornPA, Dourado ME, Hughes RAC, Islam B, Kusunoki S, Pardo CA, Reisin R, Sejvar JJ,Shahrizaila N, Soares C, Umapathi T, Wang Y, Yiu EM, Willison HJ, Jacobs BC. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. 2019 Nov;15(11):671-683.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






我初中时得过这种病
87
学习了,很有帮助,感谢作者的辛苦
82
学习了,感谢
87
学习了,总结的很好
92
#格林-巴利综合征#
83
#综合征#
82
学习
103
收藏
97
学习了,谢谢分享
101