病例报告:一例表现为噬血细胞性淋巴组织细胞增多症的Wolman病
2023-08-30 田医生 MedSci原创 发表于上海

溶酶体贮积病是由溶酶体酶或蛋白质缺陷引起的一系列疾病,进而导致人体内蛋白质和脂质无法降解及其在不同部位的积累,引起各种疾病的表现。
噬血细胞性淋巴组织细胞增多症(Hemophagocytic lymphohistiocytosis,HLH)是一种危及生命的病理性免疫激活综合征,主要发生在儿童中,其发病机制源于免疫稳态失调。活化的淋巴细胞和巨噬细胞不受控制的增殖会引起剧烈的炎症反应,引发与该疾病相关的症状和体征。
发热、肝脾肿大、全血细胞减少、高甘油三酯血症和/或低纤维蛋白原血症、铁蛋白升高、自然杀伤(NK)细胞活性缺失、可溶性CD25量增加是HLH的特征。
HLH分为家族性和继发性。原发性、先天性或家族性HLH主要发生在婴儿中,由PRF1、MUNC134和STX11等基因突变引起。继发型HLH与感染、恶性肿瘤、免疫缺陷、风湿病和代谢性疾病相关,包括戈谢病、赖氨酸尿蛋白不耐受症和溶酶体酶缺乏症等。
原发性HLH主要通过类固醇和化疗进行免疫抑制治疗,同时也可以通过异体造血干细胞移植进行治疗。继发性HLH的治疗应首先治疗潜在疾病,如感染等。
溶酶体贮积病是由溶酶体酶或蛋白质缺陷引起的一系列疾病,导致人体内蛋白质和脂质无法降解及其在不同部位的积累,引起各种疾病的表现,如尼曼-皮克病、粘多糖病、庞贝病、胆固醇酯贮积病和Wolman病(Wolman disease,WD)。
WD很罕见,在10万例新生儿中不到1例。它是由LIPA基因突变引起的溶酶体酸性脂肪酶(lysosomal acid lipase,LAL)缺失引起的。这种疾病的特点是在肝、脾、肠和淋巴结等组织的溶酶体中积聚泡沫状脂滴。患者表现为肝脾肿大、发育不良、吸收不良和器官特异性症状。肾上腺钙化是WD的独特表现。脂肪酶替代疗法为WD患者提供了更好的预后,在引入酶替代疗法之前,HSCT和肝移植被认为是有益的。
HLH和WD的临床表现和实验室结果在很大程度上重叠,这就是为什么在许多情况下,直到遗传分析结果出来才能做出明确的诊断。如果患者有继发性HLH的征象时,WD是一种潜在疾病可能,那么及时使用酶替代治疗可能会导致更高的生存率。
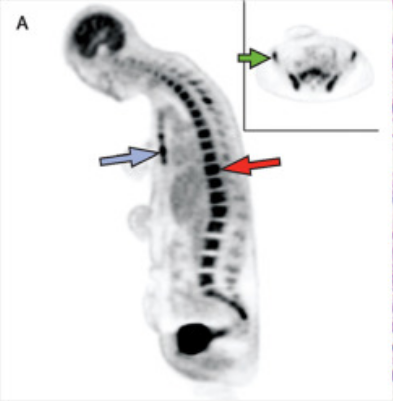
国外研究团队报告了一例4.5个月大的男孩,因接种疫苗后发热2周、黄疸和嗜睡5天而入院,无呕吐、腹泻,无抽搐或任何其他神经系统症状。父母诉患儿从出生起就有明显的腹胀。经体格检查,发现患儿面色苍白,黄疸,有轻度呼吸窘迫。腹部明显肿胀,触诊脾、肝肿大。未见皮疹及淋巴结改变。
入院时患儿凝血功能异常,肝酶升高,高直接胆红素血症,白蛋白低,铁蛋白高。患者入院时存在贫血,入院时正常的血小板、中性粒细胞、淋巴细胞在复查后逐渐下降。
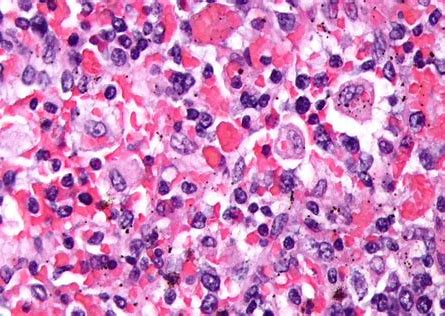
初步疑诊为HLH,开始进行免疫抑制治疗。不幸的是,患儿在入院17天后死亡,临床表现为肝衰竭导致的心肺功能受损。
WD是在孩子死后被诊断出来的,遗传学结果回报显示患儿LIPA基因中引入了一个新的突变:外显子4:NM_001127605: c. G353A (p.G118D),将甘氨酸转化为天冬氨酸,遗传分析结果与WD一致。
既往文献报告过有一些病例最初诊断为噬血细胞性淋巴组织细胞增多症的WD患者,考虑到WD与噬血细胞性淋巴组织细胞增多症的表现相似,应当对疑似噬血细胞性淋巴组织细胞增多症患者的表现予以特别关注,并及时进行适当的治疗,可以挽救患者的生命。
参考文献:
Asna Ashari K, Azari-Yam A, Shahrooei M, Ziaee V. Wolman disease presenting with hemophagocytic lymphohistiocytosis syndrome and a novel LIPA gene variant: a case report and review of the literature. J Med Case Rep. 2023 Aug 29;17(1):369. doi: 10.1186/s13256-023-04116-4. PMID: 37641143.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言