[摘要] 染色体碎裂是在应激(电离辐射、化学刺激、感染等)刺激下一条或几条染色体局部区域发生碎裂,然后以随机顺序组合在一起的复杂性基因组重排事件。近年来,通过全基因组测序、单核苷酸多态性阵列、比较基因组杂交等技术发现,这种高度复杂的基因组畸变事件普遍存在于各类型的肿瘤中,在肉瘤和神经系统肿瘤中的发生频率较高。发生染色体碎裂的机制尚不清楚,目前已知有两种主要的染色体碎裂形成机制:① 微核的形成及微核内染色体的碎裂;② 断裂-融合-桥循环的产生。染色体碎裂对于绝大多数细胞来说是灾难性的,但仍有部分细胞在经历此过程后存活下来,细胞内碎裂的染色体片段通过非同源末端连接、交替端部连接及微同源介导的断裂诱导复制等修复方式随机组合使基因组发生重排,重排后的基因组可能出现癌基因的扩增、抑癌基因的删除或者形成融合基因,赋予细胞显著的选择性生长优势,推动肿瘤的发生、发展。近年来的临床队列研究显示,肿瘤患者的临床预后也与染色体碎裂发生频率有关。染色体碎裂发生的频率越高,患者的预后越差。肿瘤中发生染色体碎裂的普遍性以及较差的预后提示检测染色体碎裂可能是评估肿瘤患者生存及预后的一种方式。已有研究表明,染色体碎裂在肿瘤的发生、发展过程中起着重要作用,但目前有关染色体碎裂现象的原因、导致肿瘤发生的机制、染色体碎裂的诊断及治疗等问题仍有待深入研究。随着对染色体碎裂相关问题的解答,将有助于更好地理解染色体碎裂在肿瘤进展中的作用,为肿瘤的预防及治疗提供新的理论依据。本文就染色体碎裂与肿瘤发生研究进展进行综述。
[关键词] 染色体碎裂;基因组重排;肿瘤发生
[Abstract] Chromothripsis is a complex genomic rearrangement event in which localized regions of one or several chromosomes fragment in response to stress (ionizing radiation, chemical stimulation, infection, etc.) and then assembles in random order. In recent years, through whole genome sequencing, single nucleotide polymorphism array, comparative genome hybridization and other technologies, it has been found that such highly complex genomic aberration events are common in all types of tumors, and they occur more frequently in sarcoma and nervous system tumors. The mechanism of chromothripsis is not clear, and there are two main mechanisms of chromothripsis formation: ① Formation of micronuclei and fragmentation of chromosomes in micronuclei; ② Generation of breakage-fusion-bridge cycles. Chromothripsis is catastrophic for the vast majority of cells, but some cells still survive this process. The fragmented chromosome fragments in cells are randomly combined to cause genome rearrangement through non-homologous end-joining, alternative end joining and microhomology-mediated break-induced replication. Oncogene amplification, tumor suppressor gene deletion or fusion gene formation may occur in the rearranged genome, which endowing cells with significant selective growth advantages and promoting the occurrence and development of tumors. Some criteria have been proposed to infer whether chromothripsis occurs in the genome. Recent clinical cohort studies have shown that the clinical prognosis of cancer patients is also related to the frequency of chromothripsis. The higher the frequency of chromothripsis, the worse the prognosis. The prevalence and poor prognosis of chromothripsis in tumors suggest that detection of chromothripsis may be a way to evaluate the survival and prognosis of cancer patients. It has been found that chromothripsis plays an important role in the occurrence and development of tumors, however, the causes of chromothripsis, the mechanism of tumorigenesis, the diagnosis and treatment of chromothripsis remain to be further studied. With the answers to the related questions of chromothripsis, it will be helpful to better understand the role of chromothripsis in tumor progression, and provide a new theoretical basis for the prevention and treatment of cancer. This article reviewed the research progress of chromothripsis and tumorigenesis.
[Key words] Chromothripsis; Genomic rearrangement; Tumorigenesis
当细胞受到物理辐射、化学刺激或感染等应激刺激后,细胞内的染色体在某个局限的区域内发生染色体碎裂(chromothripsis),发生复杂重排[1],染色体碎裂通常局限于一个或几个染色体的有限区域,在有限的区间内短时间碎裂为数十个到数百个染色体片段,并发生随机的重新排列,导致重排的染色体片段基因拷贝数的复杂变化(图1)。染色体碎裂最初是在一名淋巴细胞白血病患者基因组中发现的,高通量测序显示该患者基因组存在42个基因位点的重排,但这些重排集中位于4号染色体长臂上[2]。这个发现与渐进式遗传事件作为致癌的原因相反,这种短时间、小范围的频繁染色体重排导致的肿瘤发生并不是逐渐积累的[3],研究人员把这种染色体重排定义为染色体碎裂。为区分染色体碎裂与其他基因组重排,Korbel等[4]提出了染色体碎裂的主要特征:① 染色体碎裂引起基因重排的染色体断裂点(break points)富集在小范围区域,通常在50 kb内可以观察到5~10个染色体断裂点;② 染色体碎裂形成的基因片段重排时其连接方向和顺序具有随机性;③ 染色体碎裂通常发生在亲本染色体中两条同源染色体中的一条上;④ 发生染色体碎裂的区域所在基因杂合性缺失(loss of heterozygosity,LOH)和保留交替出现;⑤ 发生染色体碎裂的区域所在基因的拷贝数不稳定,变化复杂;⑥ 通过断点连接每个片段要么保留在新生染色体中,要么从新生染色体中丢失。随着对肿瘤发生、发展研究的不断深入,人们发现染色体碎裂在诸多肿瘤中普遍存在,研究染色体碎裂对阐述肿瘤发生的机制、探索肿瘤发展的过程及治疗肿瘤等诸多方面均起着十分重要的作用。近年来对染色体碎裂的研究进展迅速,本文就染色体碎裂现象在肿瘤发生、发展中的作用及可能的形成机制进行综述。
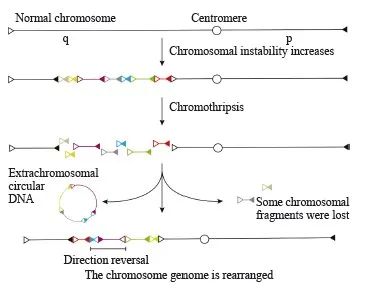
图1 染色体碎裂示意图
Fig. 1 Schematic diagram of chromothripsis
1 染色体碎裂在肿瘤发生、发展中的作用
染色体碎裂对于真核细胞来说是灾难性的,但仍有部分细胞能够在经历染色体碎裂事件后存活下来。由于染色体碎裂,细胞内含有高度突变的基因组图谱,在某种程度上导致了肿瘤的发生;而在肿瘤细胞内发生的染色体碎裂可能赋予了肿瘤细胞显著的选择性生长优势,从而促进肿瘤进展。
染色体碎裂对于不同肿瘤细胞的影响是不同的。为探究染色体碎裂在肿瘤中的发生频率,有研究[5]通过对全基因组泛癌分析(pan-cancer analysis of whole genome,PCAWG)中的38种肿瘤类型的2 658个全癌基因组及其匹配的正常组织进行整合分析,在其中587例样本(22.3%)中发现了染色体碎裂事件,且最常见于肉瘤、胶质母细胞瘤、肺鳞状细胞癌、黑色素瘤和乳腺癌。Stephens等[2]利用单核苷酸多态性阵列(single nucleotide polymorphism array,SNP array)分析了746种肿瘤细胞系,获得了肿瘤细胞系的高分辨率拷贝数图谱,他们的研究发现染色体碎裂在肿瘤细胞系中较为常见,总体发生率为2%~3%,不同肿瘤类型发生频率不同,最常发生在黑色素瘤(4/746)及小细胞肺癌(3/746)细胞系中。Voronina等[6]进一步研究了在所有类型的肿瘤组织中染色体碎裂发生的频率,研究人员总共分析了28种肿瘤,包括634个肿瘤组织的高通量测序(316个全基因组测序数据和318个全外显子组测序数据),发现染色体碎裂在所有病例的发生率为49%,提示染色体碎裂可能在多种肿瘤中起着重要作用,同时研究人员发现染色体碎裂在恶性外周神经鞘瘤中最常见(6/6)。Cortés-Ciriano等[7]利用全基因组测序技术分析了38种肿瘤共2 658个肿瘤组织,发现染色体碎裂事件在肿瘤中普遍存在,但在不同肿瘤中染色体碎裂的发生频率差异较大,其中脂肪肉瘤(100.0%)和骨肉瘤(77.0%)中染色体碎裂发生频率高,相比之下,甲状腺腺癌(3.3%)、B细胞慢性淋巴细胞白血病(1.2%)和毛细胞星形细胞瘤(0.0%)几乎不发生染色体碎裂。Bolkestein等[8]使用全基因组和全外显子组测序分析了252例乳腺癌患者(149例转移性乳腺癌患者,63例未治疗的原发肿瘤患者,29例肿瘤局部复发的患者和11对纵向配对患者)的染色体碎裂现象,发现染色体碎裂现象影响了相当比例的乳腺癌患者。同时还发现了与转移性乳腺癌染色体碎裂显著相关的信号转导通路,如SRC、MYC、mTOR和ATM,其中一些信号转导通路也与其他肿瘤发生染色体碎裂有关。Rustad等[9]通过全基因组测序对752例多发性骨髓瘤患者的队列进行了结构变异(structural variants,SV)分析。他们发现了68个涉及17个新候选驱动基因的SV热点,包括治疗靶点BCMA(TNFRSF17)、SLAMF和MCL1;并在24%的患者中发现了染色体碎裂,且与不良的临床结果独立相关。但是由于用来评估染色体碎裂的数据类型不同,或者对染色体碎裂的定义和标准不同,又或者是评分方式不同的原因,使得不同研究人员观察到染色体碎裂在肿瘤中的发生率不同[6]。染色体碎裂可能是导致肿瘤发生、发展的因素之一,深入研究染色体碎裂的机制及原因,可能为治疗肿瘤提供新的理论依据。以下是近年来染色体碎裂在肿瘤发生、发展中的机制研究。
1.1 染色体碎裂导致基因的扩增及缺失
碎裂的染色体在重组后可以导致基因的扩增或缺失,在不同肿瘤类型中发生不同的重组模式和频率。研究[5]发现,在脂肪肉瘤中,染色体碎裂事件通常涉及多个染色体,导致基因组MDM2扩增或与TERT扩增;在胶质母细胞瘤中,染色体碎裂倾向于影响离端粒较远的单个染色体上的较小区域,导致EGFR和MDM2的局部扩增及CDKN2A缺失;在肢端型黑色素瘤中,染色体碎裂常导致CCND1扩增;而在肺鳞状细胞癌中,染色体碎裂常导致SOX2扩增。
研究人员在发生基因扩增的肿瘤样本中发现了染色体外DNA(extrachromosomal DNA,ecDNA),推测造成基因扩增的原因可能与碎裂的染色体组装成染色体外DNA有关[2,10]。有研究[11]发现,ecDNA的结构为缺少着丝粒和端粒的环状DNA,即染色体外环状DNA (extrachromosomal circular DNA,eccDNA),并具有自我复制的功能。Kim等[12]利用全基因组测序数据分析3 212例患者肿瘤样本和1 810个非肿瘤样本,在大多数肿瘤样本中均检测到了大量成对的ecDNA存在,研究人员将这种成对的ecDNA命名为双微体(double minute,DM),但DM在血液和正常组织中并不常见。随着研究进展,Turner等[13]发现双微体只是eccDNA中的一种形式。Turner等[13]在肿瘤细胞中发现只有30%的eccDNA为DM,eccDNA能以单体及双微体的形式存在。eccDNA在细胞分裂后会随机分布在子细胞中,使得子细胞中某些基因的拷贝数异常增加。为了研究eccDNA如何发生基因扩增,Shoshani等[11]对克隆细胞分离株进行全基因组测序,发现染色体碎裂通过依赖于多腺苷二磷酸核糖聚合酶[poly(ADP-ribose)polymerase,PARP]和DNA依赖性蛋白激酶催化亚基(catalytic subunit of DNA-dependent protein kinase,DNA-PKcs)的机制成为ecDNA扩增的主要驱动力。Rosswog等[14]对儿童成神经细胞瘤进行全基因组测序,发现连续发生的染色体碎裂和环形重组导致了很大一部分人类恶性肿瘤中的癌基因扩增和表达。如果扩增的基因行使癌基因相关的功能,那么可能导致细胞获得选择性生长优势。Stephens等[2]利用单核苷酸多态性阵列研究小细胞肺癌细胞系的基因组特征,发现在小细胞肺癌细胞中广泛存在染色体碎裂现象,染色体碎裂导致了细胞内eccDNA含有拷贝数扩增MYC基因。同时Vogt等[15]利用荧光原位杂交(flurescence in situ hybridization,FISH)染色及全基因组测序重点研究了1例异种移植的人少突胶质细胞瘤,发现其存在染色体碎裂,eccDNA中同时存在EGFR和MYC基因的扩增。这些研究提示MYC在肿瘤中的作用与eccDNA的生物学功能有十分重要的联系。Zhao等[16]在胃贲门腺癌(adenocarcinoma of gastric cardia,GCA)患者样本中发现了频繁的局灶性扩增和ecDNA,并发现GCA队列中的局灶性扩增与染色体碎裂过程有关;此外还观察到癌基因局灶性扩增的存在与预后之间存在不同的相关性,其中ERBB2局灶性扩增与预后呈正相关,而EGFR局灶性扩增与预后呈负相关。
染色体碎裂也可导致基因的缺失,抑癌基因由于染色体碎裂缺失,可能导致肿瘤的发生及促进肿瘤发展。Stephens等[2]发现染色体碎裂导致抑癌基因CDKN2A、FBXW7及WRN的丢失与脊索瘤的产生有关。Cortés-Ciriano等[7]发现染色体碎裂导致了肿瘤抑制基因缺失(2.1%)和DNA修复基因缺失(1.9%),其中包括PTEN(12/358)、TP53(8/614)、BRCA1(8/154)及BRCA2(7/270)。在发生染色体碎裂的转移性结直肠癌(metastatic colorectal cancer,mCRC)患者中,Skuja等[17]发现超过一半的患者被鉴定出8个肿瘤抑制基因(ROBO2,CADM2、FAT4、PCDH10、PCDH18、CDH18、TSG1和CTNNA3)缺失和4个癌基因(CDH12、GPM6A、ADAM29和COL11A1)缺失。他们还在70%的患者中检测到COL11A1的缺失,在进展时间较长的患者中观察到miR-1269、miR-4465、miR-1261和miR-4490的缺失。Lei等[18]利用DNA测序研究了因为染色体碎裂而发生t(8;18)(q22;q21)染色体间易位的患者,发现患者的8号和18号染色体明显破碎成了19个片段,然后以随机的顺序和方向重新连接成新生染色体,并且观察到两个疾病相关基因RAD21和EXT1因染色体碎裂而丢失。染色体碎裂对于原有基因的作用具有双面性,既可能导致基因拷贝数的扩增,也可以导致基因拷贝数的丢失,染色体碎裂有时可能导致肿瘤向更加恶性的方向进展,反之,若癌基因的拷贝数丢失而抑癌基因的拷贝数扩增,也可能抑制肿瘤的生长。肿瘤中染色体碎裂产生的eccDNA的生物学作用目前尚不明确,有待进一步深入研究。
1.2 染色体碎裂形成融合基因
在基因重组的过程中,染色体片段的随机连接,除了会导致基因的扩增或缺失外,也可能会导致新的融合基因产生。EWSR-FL1(尤文肉瘤)、SX-SS12(滑膜肉瘤)均是比较经典的融合基因[19],这些融合基因转录形成前所未有的融合蛋白,进而成为肿瘤驱动的主要因素。染色体碎裂不仅可以导致原有基因的扩增或重排,为原本沉默的基因添加可被广泛激活的启动子元件,或是产生新的融合基因编码的融合蛋白,导致肿瘤的发生或促进肿瘤发展。Ah-Pine等[20]使用全基因组单核苷酸多态性阵列对170个IDH野生型胶质母细胞瘤进行了染色体碎裂模式筛查。通过对具有染色体碎裂特征的52例胶质母细胞瘤进行RNA测序,以鉴定染色体碎裂区域内的基因融合。观察到40个肿瘤组织(40/52,77%)在染色体碎裂区域内至少具有1个基因融合。然后他们鉴定了120个候选融合基因,发现30个融合基因具有潜在的致癌活性。Northcott等[21]在髓母细胞瘤的研究中,发现了1 087个独特的髓母细胞瘤中的体细胞拷贝数畸变(somatic copy number aberration,SCNA),其中最常见的为PVT1的反复易位。PVT1是非编码RNA基因,其与MYC的形成的新的融合基因(PVT1-MYC),可导致MYC的转录激活,促进肿瘤进展,同时该研究也发现了PVT1与NDRG1形成融合基因(PVT1-NDRG1)以及小范围的串联重复导致SNCAIP拷贝数扩增。Lee等[22]为研究非吸烟者中肺腺癌的发病原因相关的突变特征,分析了138个肺腺癌全基因组测序结果,发现非吸烟患者发生肺腺癌的驱动因素往往是复杂的染色体重排,例如染色体碎裂,这种复杂的染色体重排方式产生了许多融合基因,其中74%是已知的,例如EML4-ALK、CD74-ROS1和KIF5B-RET,同时该研究也发现融合基因驱动的肺腺癌具有频繁的SETD2突变,与EGFR突变导致的肺腺癌有着不同的基因组特征,后者往往存在频繁的全基因组重复序列及TP53突变。Zhong等[23]对先天性胶质母细胞瘤中PPP1CB-ALK融合基因及其扩增进行了基因组特征分析,发现PPP1CB-ALK是由PPP1CB的外显子1~5和ALK的外显子20~29组成。他们通过对基因组进行实时荧光定量聚合酶链反应(real-time fluorescence quantitative polymerase chain reaction,RTFQ-PCR)和Sanger测序确定了基因组易位断点。并且使用下一代测序、RTFQ-PCR和FISH分析证明了PPP1CB-ALK扩增。对PPP1CB和ALK之间的基因拷贝数进行分析,认为PPP1CB-ALK融合基因可能是局部染色体碎裂后的结果。Parker等[24]为研究幕上室管膜瘤(supratentorial ependymomas)的发生机制,对41个幕上室管膜瘤的肿瘤组织进行全基因组测序,发现染色体碎裂在幕上室管膜瘤中十分常见,并且在2/3以上的幕上室管膜瘤肿瘤组织中,发现了因染色体碎裂导致的新的融合基因C11orf95-RELA,这种融合基因是肿瘤特异性的,其转录生成的C11orf95-RELA融合蛋白可以进入细胞核激活NF-κB信号转导通路,在神经干细胞导入该融合基因可以使神经干细胞转化为幕上室管膜瘤肿瘤细胞,并在小鼠体内形成肿瘤。染色体碎裂导致融合基因的概率相对较小,但由此产生的具有功能的融合基因会使细胞生长出现根本性的改变,导致肿瘤的发生、发展,近年来,染色体碎裂导致融合基因成为相关研究领域的热点,随着这些融合基因的发现,很多驱动因素十分复杂的肿瘤的发生机制得以阐述,为后续肿瘤的治疗等研究提供了理论依据。染色体碎裂导致癌基因的扩增,抑癌基因的删除和融合基因的形成会使细胞朝着不同的方向发展,同时也会使肿瘤患者获得不同的临床预后。
2 染色体碎裂对肿瘤患者临床预后的影响
染色体碎裂在肿瘤的发生、发展中起着重要作用,近来的临床队列研究显示,患者的临床预后也与染色体碎裂发生频率有关。染色体碎裂发生的频率越高,患者的预后越差。Magrangeas等[25]对764例多发性骨髓瘤(multiple myeloma,MM)患者的临床随访结果显示,1.3%的患者(10例)存在高频染色体碎裂,这些患者中50%(5/10)在诊断后10个月内迅速复发,其中4例死亡(3例患者死亡发生在复发后12个月内),这与大多数MM的发生、发展速度不符,提示染色体碎裂象可能导致MM的恶性程度改变,具有染色体碎裂特征的MM患者预后不良。为了探索基因组不稳定性和染色体碎裂与临床预后的相关性,Hirsch等[26]利用阵列比较基因组杂交(array comparative genomic hybridization,aCGH)对20例黑色素瘤患者进行了研究,通过比较10例确诊后死于黑色素瘤的患者(中位生存期为3.7年)和10例存活的黑色素瘤患者(中位无病生存期为14.8年)的基因组特征,发现预后较差的黑色素瘤患者(平均每例发生13.9次染色体结构改变)比预后良好的患者(平均每例发生1.6次染色体结构改变)有着更高的基因组不稳定性和局灶性拷贝数变化。并且在预后较差的患者中有两例拷贝数的变化符合染色体碎裂的标准。这些研究提示检测染色体碎裂可能是评估肿瘤患者生存及预后的一种方式,这也印证了高频发生染色体碎裂的肿瘤类型,患者的生存相对较差。
3 染色体碎裂形成的可能机制
染色体碎裂的形成主要发生在真核细胞有丝分裂过程中。当细胞接收到有丝分裂的信号时,细胞核中的染色质开始浓缩,在形成染色体的过程中,如果暴露在较强的应激刺激下(如电离辐射[27]、化学刺激[28]、感染[29]等),会导致染色体的某个局部区域或多个邻近空间的染色体受到破坏,导致染色体区域片段的DNA损伤,包括碱基损伤、单链断裂及双链断裂。在损伤修复的过程中,碎裂的染色体片段主要通过非同源末端连接(non-homologous end-joining,NHEJ)修复在一起,交替端部连接(alternative end joining,alt-EJ)及微同源介导的断裂诱导复制(microhomology-mediated break-induced replication,MMBIR)也参与了断裂位置的损伤修复[7]。如果损伤无法修复并且十分严重,细胞接收到信号发生细胞程序性死亡(programmed cell death);但若损伤程度仍在细胞存活的接受范围内,程序性死亡的过程将终止,此时通过NHEJ随机连接的损伤DNA片段被保留下来,导致染色体碎裂的发生。以下为两种主要的染色体碎裂形成的机制。
3.1 微核的形成及微核内染色体的碎裂
微核(micronuclei,MN)是由核膜包裹染色体所组成的结构,在空间上与主核(primary nucleus)分离。微核与染色体不稳定性、基因组重排和突变有关。有丝分裂时滞后分裂的染色体、两端没有着丝粒牵引的染色体以及发生分裂错误的染色体在有丝分裂中未被纺锤体牵拉至细胞两级,停留在赤道板附近然后随机分离至子细胞的细胞质中,被细胞核外形成的核膜包裹,形成微核,微核中通常含有一条或几条染色体[30-33]。目前已知有两种微核导致染色体碎裂的机制。由于微核与原细胞核在核膜组成上的差异,使得微核核膜不稳定从而容易发生破裂,暴露的微核内染色体与细胞质某些蛋白接触,导致染色体碎裂。研究发现微核不能运输并维持核膜和基因组完整性所必需的关键蛋白质,并且纺锤体微管阻断了核孔复合物(nuclear pore complex,NPC)和其他膜蛋白在滞后染色体上的组装,导致核包膜组装中不可逆的缺陷[34]。Vietri等[35]发现,由于微核体积小,缺乏初生核限制CHMP7-LEMD2积累的能力,CHMP7-LEMD2是一种检测细胞核完整性的传感器。膜蛋白复合物Ⅲ(endosomal sorting complexes required for transport Ⅲ,ESCRT-Ⅲ)在核膜的活性由CHMP7亚基协调。ESCRT-Ⅲ可以确保核膜在有丝分裂晚期重新封闭,并在间期修复。因此,ESCRT-Ⅲ活性可能对维持核膜包围的其他基因组结构的完整性至关重要。但是ESCRT-Ⅲ的异常积累将会导致广泛的膜变形、DNA损伤和染色体断裂。Mammel等[36]证明了微核染色体的长度和基因密度,是维持微核膜稳定性的关键因素并决定了微核破裂的时间。微核大小与微核膜中的层蛋白B1水平和核孔密度密切相关,但层蛋白B1水平并不能完全预测核膜的稳定性。相反,尽管基因密度小的微核与基因密度大的微核相比层蛋白B1水平很低,但是基因密度小的微核减少了核层间隙,延长了破裂时间。作者认为核膜组成缺陷只是维持核膜稳定性的部分因素。他们提出,一个与基因密度相关的未知因素,它有一个单独的功能,即抑制核层间隙的出现,延迟核膜破裂直到细胞周期后期。微核膜破裂后,细胞质里的3′-核酸修复外切酶1(three prime repair exonuclease 1,TREX1)进入微核内损伤染色体,诱导DNA双链断裂,发生染色体碎裂,Mohr等[37]纯化微核后发现内质网(endoplasmic reticulum,ER)相关的核酸酶TREX1通过降解微核包膜破裂后暴露的微核DNA,从而抑制环状GMP-AMP合酶(cyclic GMP-AMP synthase,cGAS)的激活,阻止细胞免疫应答。实验证明,ER可以进入破裂的微核,并在TREX1溶解DNA中发挥关键作用。TREX1基因的突变,可以解除TREX1与ER的连接,破坏TREX1基因定位到微核,继而减少微核DNA损伤,并增强cGAS的激活。此外,微核内部的许多生物学过程(DNA转录和复制、DNA损伤修复等)相比于细胞主核(primary nucleus)缺失或滞后,表现出异步的复制或者复制不足。当细胞发出有丝分裂的信号时,微核内染色质的复制尚未完成,此时过早发生了其向染色体的转变,也可能导致染色体不成熟而发生碎裂,也有学者提出染色质过早凝聚(premature chromosome condensation,PCC)可能是微核染色体碎裂的原因。Pantelias等[3]使用药物RO-3306(一种促进G2/M边界处细胞周期停滞的CDK1选择性ATP竞争性抑制剂)来控制主核和微核之间的细胞周期异步程度,通过延迟主核进入有丝分裂,使得微核获得额外的时间来完成DNA复制。他们在γ射线照射人淋巴细胞产生的异步微核细胞和细胞融合产生的异步多核中国仓鼠卵巢(Chinese hamster ovary,CHO)细胞中进行了细胞分裂间期遗传学分析。研究结果表明,有丝分裂过程中微核细胞或多核细胞的异步染色质过早凝聚动力学是染色体破碎的决定性因素,可能是染色体断裂的重要机制。微核可以在细胞分裂后随机进入子细胞。Vázquez-Diez等[38]发现微核内部的染色体着丝粒功能受损,提出微核内染色体即使可能存在着丝粒,也无法进行正常的有丝分裂,子代的微核是由亲本细胞遗传的,同一个微核可以遗传多次,而不需要重新并入主核,也不会改变细胞分裂的动力学。
细胞质核酸酶TREX1的降解作用或染色质过早凝聚被认为是造成微核内染色体发生碎裂的主要机制。碎裂后的染色体片段以随机方式发生重组,在下一次分裂时可以重新并入主核[39],成为基因组不稳定的来源。
3.2 断裂-融合-桥循环(breakage-fusion-bridge cycles,BFB)的产生
当编码端粒酶的基因发生突变或缺失后,细胞受到外界刺激,在氧化应激、化学物质、离子辐射等作用下,端粒发生损伤无法修复,端粒失去对染色体末端的保护。细胞内两条没有端粒保护的染色单体会发生损伤修复融合成双着丝粒染色体(dicentric-chromosome),在有丝分裂过程中,纺锤体牵引双着丝粒染色体,形成染色体桥(chromosome bridge),随着纺锤体的牵拉,染色体桥发生断裂。Umbreit等[40]通过实验发现BFB循环中染色体桥的断裂需要机械力的牵拉作用;但也有学者提出染色体桥的断裂是由于核膜破裂时相关酶的作用, Maciejowski等[41]利用体外端粒危机模型研究了染色体碎裂的潜在机制,发现在TREX1缺失的情况下,端粒危机引起的基因组改变主要包括BFB循环和简单的基因组重排,而不是染色体碎裂,提示肿瘤中染色体碎裂可能是通过TREX1介导的双着丝粒染色体碎裂而发生的。
断裂的染色体片段进入子细胞由于仍然没有端粒保护,其在复制过程形成姐妹染色单体后,由于姐妹染色单体断裂端依旧会发生损伤修复融合在一起,在有丝分裂后期会形成双着丝粒染色体,但这种染色单体断裂端的融合并不一定是同源的,若两条或多条非同源染色单体的断裂端随机融合,情况将会更为复杂。不论发生哪种融合,断裂染色体融合后是殊途同归的,结果均会形成染色体桥,并因为上述机制在有丝分裂过程中断裂,这种染色体断裂、染色体融合、形成染色体桥继而发生断裂的过程,称为断裂-融合-桥循环(BFB循环),若在BFB循环中发生不对称断裂,会导致该处基因拷贝数的改变(图2)。如此往复的BFB循环最终导致的染色体重排存在着频繁的基因拷贝数改变。染色体桥发生断裂产生的染色体片段有可能形成微核。Umbreit等[40]使用活细胞成像技术来观察染色体桥,他们发现第一代细胞发生染色体桥断裂后并不会产生微核,但是52%的第二代细胞中的染色体桥断裂后出现了微核。即使第一代细胞不发生染色体桥断裂,也有65%的概率产生微核。相比之下,没有染色体桥的细胞不会产生微核。
微核的形成和BFB循环的产生可能是发生染色体碎裂的两种主要原因,不同的肿瘤类型可能由某一种机制或几种机制共同作用产生染色体碎裂事件,并且由于肿瘤异质性的原因,染色体碎裂在不同的肿瘤类型中的发生频率不同,所造成的后果也不同。
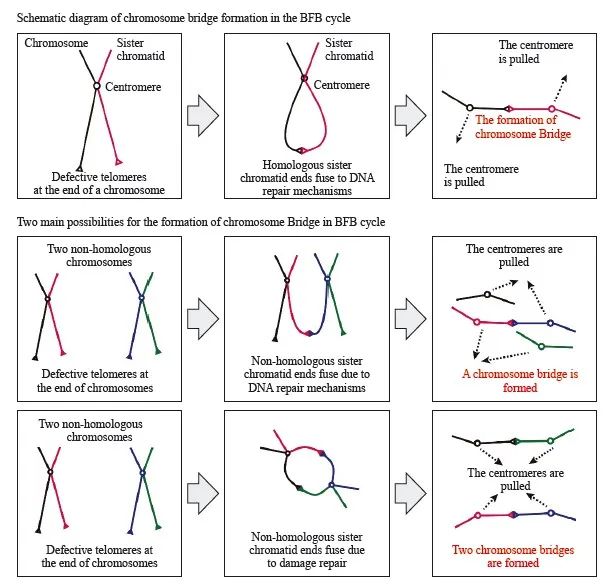
图2 BFB循环中染色体桥形成示意图
Fig. 2 Schematic diagram of chromosome bridge formation in the BFB cycle
4 总结与展望
综上所述,染色体碎裂是染色体在一次灾难性事件中染色体局部区域发生碎裂,然后碎裂片段随机连接而产生大量聚集的基因重排。发生染色体碎裂可能导致癌基因的扩增、肿瘤抑制基因或DNA损伤修复基因的缺失甚至产生新的融合基因,从而导致肿瘤的发生、发展,并影响患者预后。染色体碎裂导致基因组结构和功能发生改变,但驱动染色体碎裂的原因和机制目前尚不完全清楚,如何围绕染色体碎裂这一现象开展诊断和治疗创新型研究和药物开发仍无明确方向,但是随着这一领域的迅速发展,重点解析染色体碎裂在不同肿瘤中的作用,将会翻开肿瘤遗传学新的一页,也可能为肿瘤精准靶向治疗提供新的理论依据。
利益冲突声明:所有作者均声明不存在利益冲突。
[参考文献]
[1] HOLLAND A J, CLEVELAND D W. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements[J]. Nat Med, 2012, 18(11): 1630-1638.
[2] STEPHENS P J, GREENMAN C D, FU B Y, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development[J]. Cell, 2011, 144(1): 27-40.
[3] PANTELIAS A, KARACHRISTOU I, GEORGAKILAS A G, et al. Interphase cytogenetic analysis of micronucleated and multinucleated cells supports the premature chromosome condensation hypothesis as the mechanistic origin of chromothripsis[J]. Cancers (Basel), 2019, 11(8): 1123.
[4] KORBEL J O, CAMPBELL P J. Criteria for inference of chromothripsis in cancer genomes[J]. Cell, 2013, 152(6): 1226-1236.
[5] ICGC/TCGA PAN-CANCER ANALYSIS OF WHOLE GENOMES CONSORTIUM. Pan-cancer analysis of whole genomes[J]. Nature, 2020, 578(7793): 82-93.
[6] VORONINA N, WONG J K L, HÜBSCHMANN D, et al. The landscape of chromothripsis across adult cancer types[J]. Nat Commun, 2020, 11(1): 2320.
[7] CORTÉS-CIRIANO I, LEE J J, XI R B, et al. Comprehensive analysis of chromothripsis in 2 658 human cancers using wholegenome sequencing[J]. Nat Genet, 2020, 52(3): 331-341.
[8] B O L K E S T E I N M , WO N G J K L , THEWE S V , e t a l . Chromothripsis in human breast cancer[J]. Cancer Res, 2020, 80(22): 4918-4931.
[9] RUSTAD E H, YELLAPANTULA V D, GLODZIK D, et al. Revealing the impact of structural variants in multiple myeloma[J]. Blood Cancer Discov, 2020, 1(3): 258-273.
[10] LIAO Z Y, JIANG W, YE L Y, et al. Classification of extrachromosomal circular DNA with a focus on the role of extrachromosomal DNA (ecDNA) in tumor heterogeneity and progression[J]. Biochim Biophys Acta Rev Cancer, 2020, 1874(1): 188392.
[11] SHOSHANI O, BRUNNER S F, YAEGER R, et al. Chromothripsis drives the evolution of gene amplification in cancer[J]. Nature, 2021, 591(7848): 137-141.
[12] KIM H, NGUYEN N P, TURNER K, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers[J]. Nat Genet, 2020, 52(9): 891-897.
[13] T U R N E R K M , D E S H P A N D E V , B E Y T E R D , e t a l . Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity[J]. Nature, 2017, 543(7643): 122-125.
[14] ROSSWOG C, BARTENHAGEN C, WELTE A, et al. Chromothripsis followed by circular recombination drives oncogene amplification in human cancer[J]. Nat Genet, 2021, 53(12): 1673-1685.
[15] VOGT N, GIBAUD A, LEMOINE F, et al. Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma[J]. Nucleic Acids Res, 2014, 42(21): 13194-13205.
[16] ZHAO X K, XING P W, SONG X, et al. Focal amplifications are associated with chromothripsis events and diverse prognoses in gastric cardia adenocarcinoma[J]. Nat Commun, 2021, 12(1): 6489.
[17] SKUJA E, BUTANE D, NAKAZAWA-MIKLASEVICA M, et al. Deletions in metastatic colorectal cancer with chromothripsis[J]. Exp Oncol, 2019, 41(4): 323-327.
[18] LEI M, LIANG D S, YANG Y F, et al. Long-read DNA sequencing fully characterized chromothripsis in a patient with Langer-Giedion syndrome and Cornelia de Lange syndrome-4[J]. J Hum Genet, 2020, 65(8): 667-674.
[19] GLENFIELD C, INNAN H. Gene duplication and gene fusion are important drivers of tumourigenesis during cancer evolution[J]. Genes (Basel), 2021, 12(9): 1376.
[20] AH-PINE F, CASAS D, MENEI P, et al. RNA-sequencing of IDH-wild-type glioblastoma with chromothripsis identifies novel gene fusions with potential oncogenic properties[J]. Transl Oncol, 2021, 14(1): 100884.
[21] NORTHCOTT P A, SHIH D J, PEACOCK J, et al. Subgroupspecific structural variation across 1 000 medulloblastoma genomes[J]. Nature, 2012, 488(7409): 49-56.
[22] LEE J J, PARK S, PARK H, et al. Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma[J]. Cell, 2019, 177(7): 1842-1857.e21.
[23] ZHONG Y M, LIN F M, XU F, et al. Genomic characterization of a PPP1CB-ALK fusion with fusion gene amplification in a congenital glioblastoma[J]. Cancer Genet, 2021, 252/253: 37-42.
[24] PARKER M, MOHANKUMAR K M, PUNCHIHEWA C, et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma[J]. Nature, 2014, 506(7489): 451-455.
[25] MAGRANGEAS F, AVET-LOISEAU H, MUNSHI N C, et al. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients[J]. Blood, 2011, 118(3): 675-678.
[26] HIRSCH D, KEMMERLING R, DAVIS S, et al. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma[J]. Cancer Res, 2013, 73(5): 1454-1460.
[27] PANTELIAS A, ZAFIROPOULOS D, CHERUBINI R, et al. Interphase cytogenetic analysis of G0 lymphocytes exposed to α-particles, C-ions, and protons reveals their enhanced effectiveness for localized chromosome shattering-a critical risk for chromothripsis[J]. Cancers (Basel), 2020, 12(9): 2336.
[28] FANT C, GRANZOTTO A, MESTAS J L, et al. DNA doublestrand breaks in murine mammary tumor cells induced by combined treatment with doxorubicin and controlled stable cavitation[J]. Ultrasound Med Biol, 2021, 47(10): 2941-2957.
[29] PLEGUEZUELOS-MANZANO C, PUSCHHOF J , ROSENDAHL HUBER A, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli[J]. Nature, 2020, 580(7802): 269-273.
[30] BLACKFORD A N, STUCKI M. How cells respond to DNA breaks in mitosis[J]. Trends Biochem Sci, 2020, 45(4): 321-331.
[31] ORR B, DE SOUSA F, GOMES A M, et al. An anaphase surveillance mechanism prevents micronuclei formation from frequent chromosome segregation errors[J]. Cell Rep, 2021, 37(6): 109783.
[32] STORCHOVA Z. Consequences of mitotic failure-the penalties and the rewards[J]. Semin Cell Dev Biol, 2021, 117: 149-158.
[33] KOLB T, ERNST A. Cell-based model systems for genome instability: dissecting the mechanistic basis of chromothripsis in cancer[J]. Int J Cancer, 2021.
[34] MAMMEL A E, HATCH E M. Genome instability from nuclear catastrophe and DNA damage[J]. Semin Cell Dev Biol, 2022, 123: 131-139.
[35] V I E T R I M , S C H U L T Z S W, B E L L A N G E R A , e t a l . Unrestrained ESCRT-Ⅲ drives micronuclear catastrophe and chromosome fragmentation[J]. Nat Cell Biol, 2020, 22(7): 856-867.
[36] MAMMEL A E, HUANG H Z, GUNN A L, et al. Chromosome length and gene density contribute to micronuclear membrane stability[J]. Life Sci Alliance, 2022, 5(2): e202101210.
[37] MOHR L, TOUFEKTCHAN E, VON MORGEN P, et al. ERdirected TREX1 limits cGAS activation at micronuclei[J]. Mol Cell, 2021, 81(4): 724-738.e9.
[38] VÁZQUEZ-DIEZ C, YAMAGATA K, TRIVEDI S, et al. Micronucleus formation causes perpetual unilateral chromosome inheritance in mouse embryos[J]. Proc Natl Acad Sci U S A, 2016, 113(3): 626-631.
[39] HINTZSCHE H, HEMMANN U, POTH A, et al. Fate of micronuclei and micronucleated cell[J]. Mutat Res Rev Mutat Res, 2017, 771: 85-98.
[40] UMBREIT N T, ZHANG C Z, LYNCH L D, et al. Mechanisms generating cancer genome complexity from a single cell division error[J]. Science, 2020, 368(6488): eaba0712.
[41] MACIEJOWSKI J, CHATZIPLI A, DANANBERG A, et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis[J]. Nat Genet, 2020, 52(9): 884-890.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言