药物临床试验的不良事件管理--DIA中国年会AE上报的挑战及更佳实践分会场专家讨论荟萃
2018-06-08 DIA DIA

新药临床试验是为药物有效性和安全性提供客观的循证医学证据,药物的安全性, 风险收益比更是药监部门评审新药临床试验的关注点。自2015年我国国家药品监督管理局开展新药注册申请相关的GCP 核查工作以来,安全性事件的报告,尤其是不良事件的漏报是核查中发生频次最高的不合格项目1。为什么会存在频发的不良事件漏报,这样的漏报对药物安全性的评估会造成什么样的影响,有什么解决之道? 本界DIA年会0406分会场
新药临床试验是为药物有效性和安全性提供客观的循证医学证据,药物的安全性, 风险收益比更是药监部门评审新药临床试验的关注点。
自2015年我国国家药品监督管理局开展新药注册申请相关的GCP 核查工作以来,安全性事件的报告,尤其是不良事件的漏报是核查中发生频次最高的不合格项目1。
为什么会存在频发的不良事件漏报,这样的漏报对药物安全性的评估会造成什么样的影响,有什么解决之道? 本界DIA年会0406分会场的专家们就业界关注的问题,分别从研究者、机构、申办方的角度对不良事件(AE)的管理做了透彻的介绍和分析,并提出改进建议;更有US FDA 检查员友情客串,从检查员角度对AE 评估和报告的重要性进行了阐述。
(1)研究者对AE 评估与报告的主体责任
药物临床试验中,研究者不但要对受试者在试验过程中发生的AE 给予足够的医疗照顾,还要根据方案、申办方和当地法规/伦理委员会的要求对发生的AE进行评估、记录、报告与跟进2。
香港大学临床试验中心执行总监Henry Yau先生从临床研究中心角度分析了研究者在AE发现(identify)、记录(document)、判断(assess)、报告(report)和跟踪(follow-up)中存在缺陷的根本原因,即四个「不」(4“NOT”) ,包括不知(NOT aware)、不能(NOT capable)、不慎(NOT careful)或/和不愿(NOT willing). 从Henry的观察看,大部分研究者对GCP 关于AE 报告的要求是基本了解,即基本的awareness是有的,缺陷的根源更多的是不能、不慎和/或不愿。
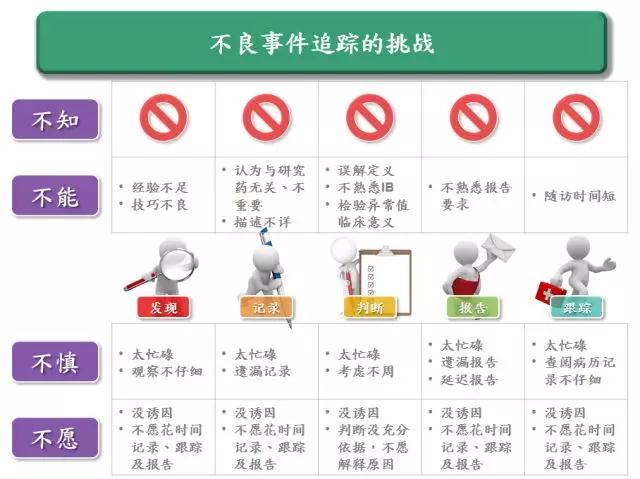
针对这些问题,Henry 也给出了解决之道,

(2) 申办者对临床试验药物安全事件的管理责任
临床试验中的AE发现、评估与报告虽来自于研究者,但申办方对所报告的安全性数据负有最终责任。与会专家一致认为,申办方对研究者的培训,过程中持续的监查和提醒、对AE数据的汇总分析以及对整个流程的质量保证能够改善AE报告的依从性。
默沙东生物统计与研究决策科学部(BARDS)亚太区负责人丁劼博士从AE数据汇总分析的角度,让与会者对AE 全流程管理有了深入的了解,从而认识到试验中完整的AE报告的重要性*。
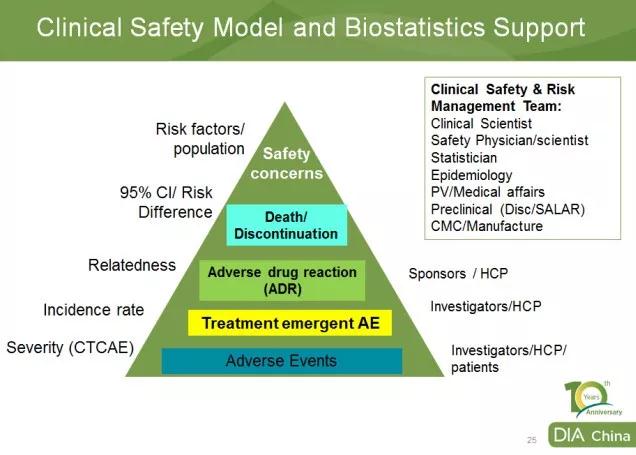
对于研究者常有的关于如何判断相关性的问题,丁博士也给出了建议,

最后,针对业界在AE报告方面普遍存在的实际问题,如SAE的发生和结束时间的采集,有临床意义的实验室异常值,以事件为疗效终点的AE报告等,专家组也开展了全面和具体的探讨。中山大学附属肿瘤医院临床试验中心机构办公室主任曹烨博士不仅介绍了自己在安全性事件管理的心得,还介绍了广东省药学会药物临床试验专业委员会的广东共识3;强生中国质量策略总监陈华女士和US FDA 资深检查员Byungja E, Marciante 分别从稽查和检查的角度阐述了AE发现,判定和报告的重要性,以及对NDA 审评的影响。
AE管理需多方合力,本着以终为始,质量源于设计的原则,申办方应在试验方案中明确AE的类型、报告时限、重大临床意义AE的考量(若适用),安全性事件的分析汇总等;在试验开始前为研究者提供研究者手册(IB)和相关培训;研究机构和PI应确保试验中的人员分配,研究者应按方案随访受试者,全面收集、审阅受试者资料,对非预期事件和实验室异常值做出判断、记录和报告;试验中,研究者或监查员对方案执行过程中的疑问应及时提出并跟进解决;申办方和机构协力加强风险管理和质量保证。
参考资料
1.《药物临床试验数据核查阶段性报告》
2.《ICH GCP》
3.《药物临床试验安全评价·广东共识》


本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#临床试验#马克
63
#DIA中国年会#
87
#荟萃#
81
#DIA#
73
#药物临床试验#
72