累趴的T细胞
● ● ●
每个人的身体内部都有一支隶属于我们个人的专属军队——免疫细胞,为保卫我们的健康冲锋陷阵,万死不辞。在众多免疫细胞中,T细胞是不可或缺的一员大将。但T细胞也有累了的时候,我们如何理解“T细胞耗竭”?
-01-
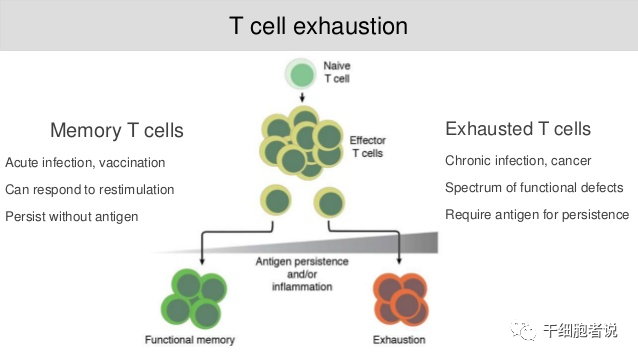
T细胞耗竭基本概念
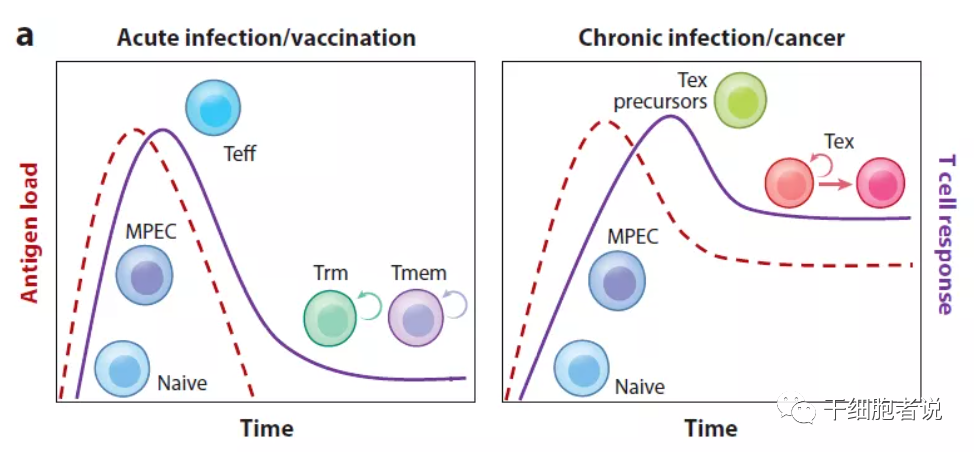
抗原的持续作用于T细胞耗竭
在慢性感染或癌症期间,由于长期暴露于持续性抗原和炎症,T细胞受到持续刺激,精疲力竭的效应T细胞逐渐失去效应功能,记忆T细胞特征也开始缺失,T细胞耗竭发生了。
T细胞是否会发生耗竭,主要受表达的抑制性受体(IRS)的水平和数量的影响,而免疫检查点抑制剂可以部分逆转T细胞耗竭状态。此外,抗原刺激的强度和感染持续时间也是重要因素。
-02-
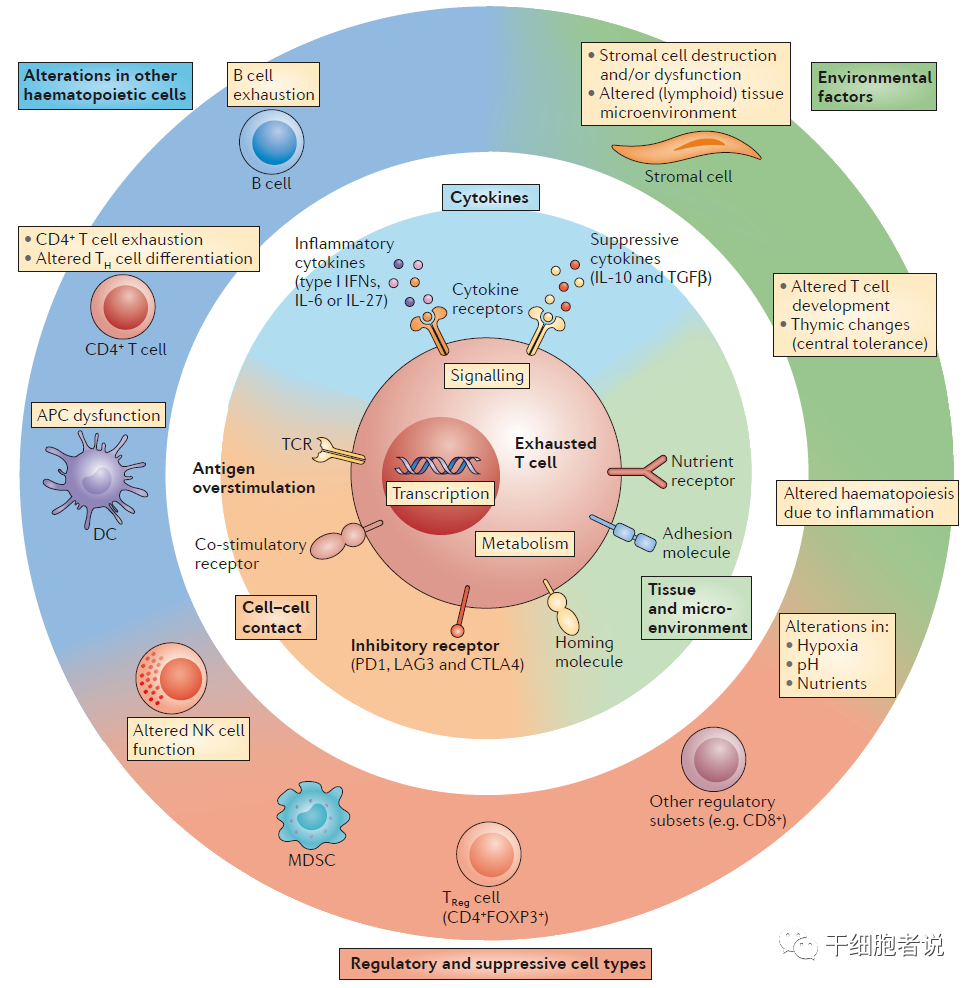
T细胞耗竭的机制概览
(1)细胞间的信号(T细胞受体接合时间延长、共刺激和/或共抑制信号)
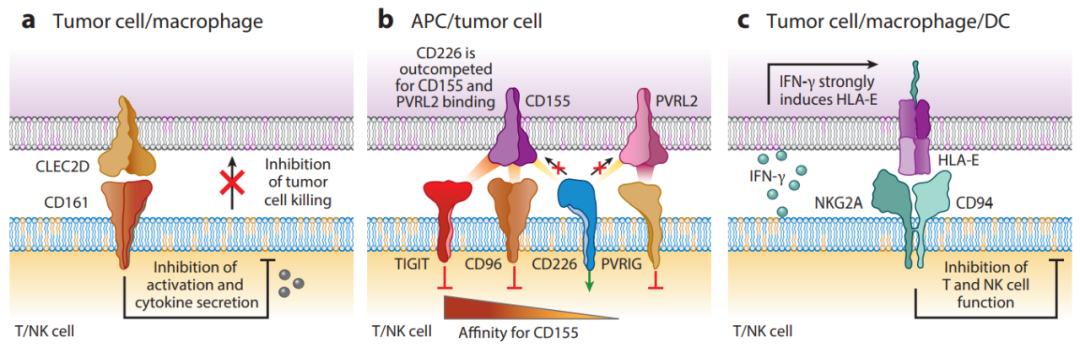
引起T细胞耗竭的原因可能是其他免疫细胞和基质细胞,除了CD8+T细胞耗竭参与已被证实,其他细胞包括:CD4+T细胞、NK细胞、抗原呈递细胞(APC)、B细胞和调节细胞(例如骨髓来源的抑制细胞<MDSC>和调节性T细胞<Treg>)。
-03-
T细胞耗竭的调控因素
慢性感染或癌症的一个关键特征:由于长期暴露于持续性抗原和炎症,T细胞受到持续刺激。效应T细胞失去效应功能,记忆T细胞特征缺失。造成T细胞耗竭主要原因是:T细胞功能缺陷(免疫异型性受体表达增加,如PD-1、CTLA4、TIM-3),可溶性免疫抑制因子(如IL-10、IL-35增加),与其他免疫细胞的互动(如Treg细胞的抑制活性),共同造成了T细胞耗竭状态。
新的证据也表明:耗竭T细胞表现出明显的表观遗传特征,这可能导致对免疫治疗的不良反应。抑制性受体表达增加是造成T细胞耗竭的主要原因,而免疫检查点抑制剂可以部分逆转T细胞耗竭状态。
▉ PD-1/PD-LI
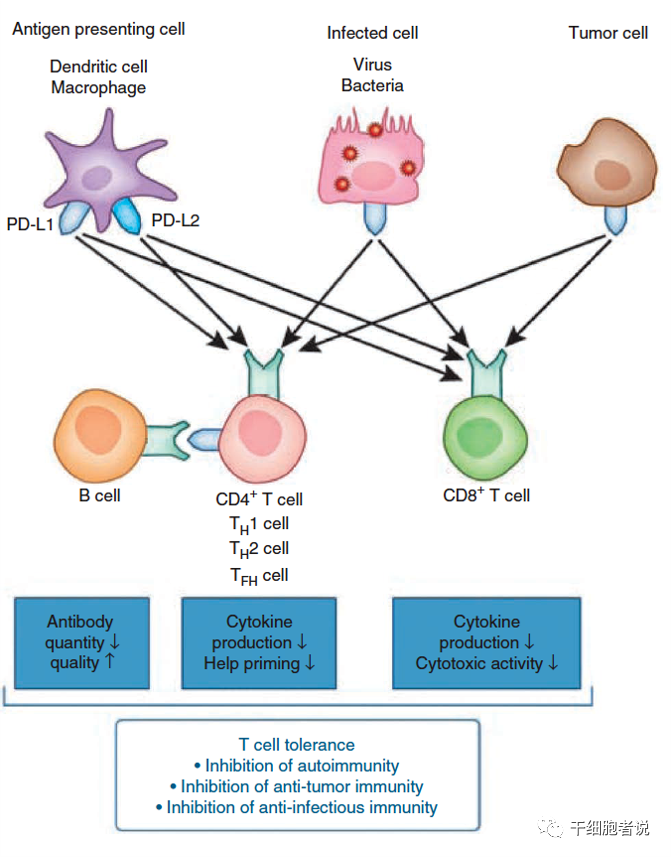
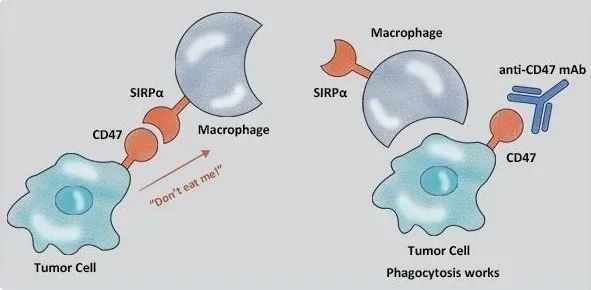
PD-1表达于 T细胞、B细胞、NK细胞、单核细胞和DC细胞表面;PD-L表达于造血细胞、非造血细胞和肿瘤细胞表面;由PD-1介导信号通路是CD8+T细胞耗竭的负向信号调控通路;
PD-1/PD-L1相互作用可诱导下游酪氨酸激酶(SYK)和磷酸酰基醇-3 激酶(PI3K)磷酸化,从而抑制下游信号传导和T细胞的生物学功能,从而促进T细胞耗竭和凋亡;促进Treg细胞增殖和免疫抑制作用,抑制NK细胞和B细胞作用。
▉ PD-1/CTLA-4
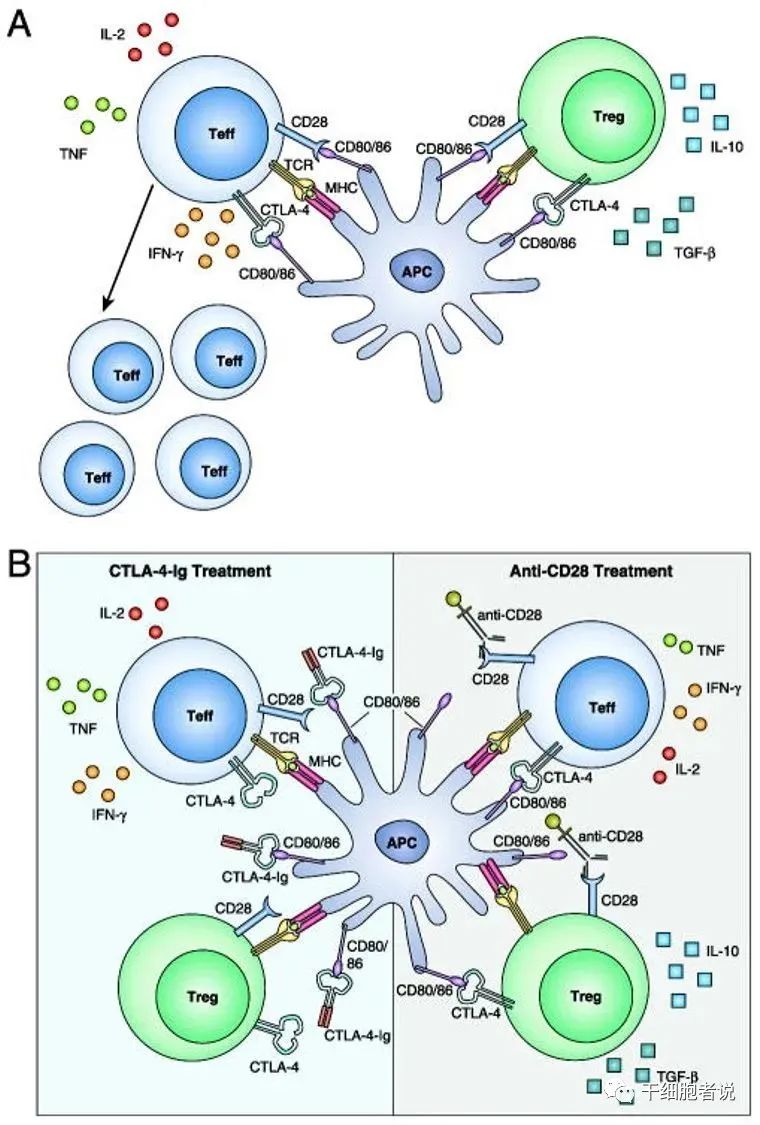
CTLA-4是CD28蛋白家族的一种抑制受体,表达于T细胞表面,可与CD28竞争性结合配体CD80/CD86发挥抑制IL-2产生的作用,从而抑制T细胞功能;CTLA-4激活磷脂酰肌醇激酶-3(PI3-K),造成T细胞功能丧失;CTLA-4结合TCR,导致条件性T细胞扩增,并将天然CD4+CD25+T细胞转为Treg,从而抑制效应T细胞。
▉ TIM-3
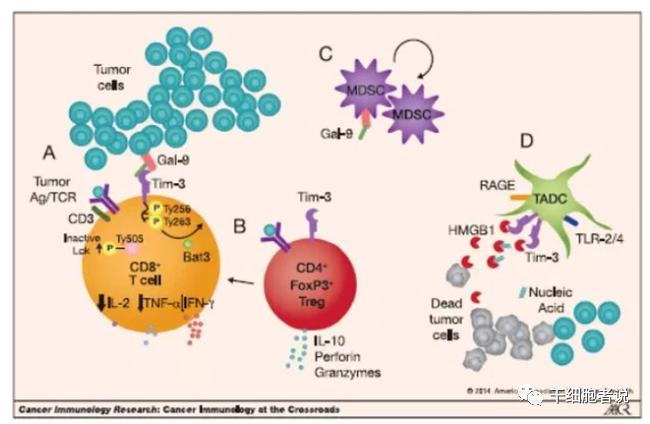
TIM-3是TIM家族成员,选择性地表达在Th1、Th17、Treg、DC、单核细胞、肥大细胞、NK、TIL等细胞;当TIM3与其配体Galectin-9 结合,可抑制Th1 和Th17 扩增,促进Th1凋亡、CD8+T细胞功能耗竭,诱导MDSC大量扩增,从而直接/间接地促进外周免疫耐受,抑制机体抗肿瘤免疫。
▉ IL-10
IL-10常与减弱T细胞活化有关,阻断IL-10可预防和/或逆转 T 细胞耗竭。DC细胞、B细胞、单核细胞、CD8+T细胞和非调节性CD4+T细胞都能产生IL-10。IL-10的作用可直接作用于T细胞,也可通过APC间接作用于T细胞,或两者兼有。
-04-
耗竭T细胞表达免疫抑制性受体,免疫检查点抑制剂可以阻断这些信号,逆转耗竭的T细胞,恢复肿瘤微环境肿瘤浸润T细胞(TIL)功能。

▉ 免疫联合疗法可增强疗效
T细胞耗竭的主要原因为免疫抑制性受体表达增加,如PD-1、CTLA-4、Tim-3等,联合受体抑制剂治疗可以增强免疫治疗效果。
 免疫联合疗法部分临床试验
免疫联合疗法部分临床试验
-05-
T细胞衰竭机制复杂,因素众多。根本原因还是抗原和炎症长期刺激 T 细胞,T细胞厌倦了这样的生活,开始躺平,不愿意搭理这些抗原或炎症,效应T细胞不再效应,记忆T细胞也不再记忆。
其实,人何尝不是如此。当你长期处于焦虑状态,体内源源不断给你提供免疫抑制剂和神经抑制剂,你的免疫系统和神经系统将持续低下(虽然我无法证实这件事 )。耗竭的T细胞就是榜样!不管怎么说,人还是要尽量开心一点,远离焦虑!
)。耗竭的T细胞就是榜样!不管怎么说,人还是要尽量开心一点,远离焦虑!

参考文献:
-
http://www.bloodjournal.org/Thorsten Zenz -
Annu. Rev. Immunol. 2019. 37:457–95 -
Nature Reviews Clinical Oncology, 2016, 13(4):202-203. -
Nat Immunol. 2013 Dec;14(12):1212-8. -
Nat Metab 2, 1001–1012(2020). -
Annu Rev Immunol. 2019 Apr 26;37:457-495.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言