
“绘”解读真报告丨基因检测如何辅助神经纤维瘤病精准诊疗?
2023-11-13 苏州绘真医学 苏州绘真医学 发表于上海

神经纤维瘤病是一种良性的周围神经疾病,属于常染色体显性遗传病。其组织学上起源于周围神经鞘神经内膜的结缔组织。它常累及起源于外胚层的器官,如神经系统、眼和皮肤等,是常见的神经皮肤综合征之一。
近日,1例4岁女童,躯干、四肢可见多发大小不等咖啡斑,双侧小脑齿状核、丘脑及苍白球、中脑多发异常信号灶,临床上考虑为神经纤维瘤病。由于没有组织样本,采集血液样本送检我司实体瘤201plus基因检测,辅助临床诊断。

图1 患者出院小结
基因检测结果提示,该患者携带NF1基因胚系杂合变异。根据《儿童及青少年神经纤维瘤病诊疗规范(2021年版)》关于神经纤维瘤病的诊断标准,初步判断该患者为1型神经纤维瘤病(NF1),以临床医生判断为最终结论。同时,检测报告提示了携带NF1胚系变异患者的遗传风险。

图2 绘真医学实体瘤201plus基因检测项目检出NF1基因胚系杂合变异
表1 神经纤维瘤病基因检测项目及其临床意义
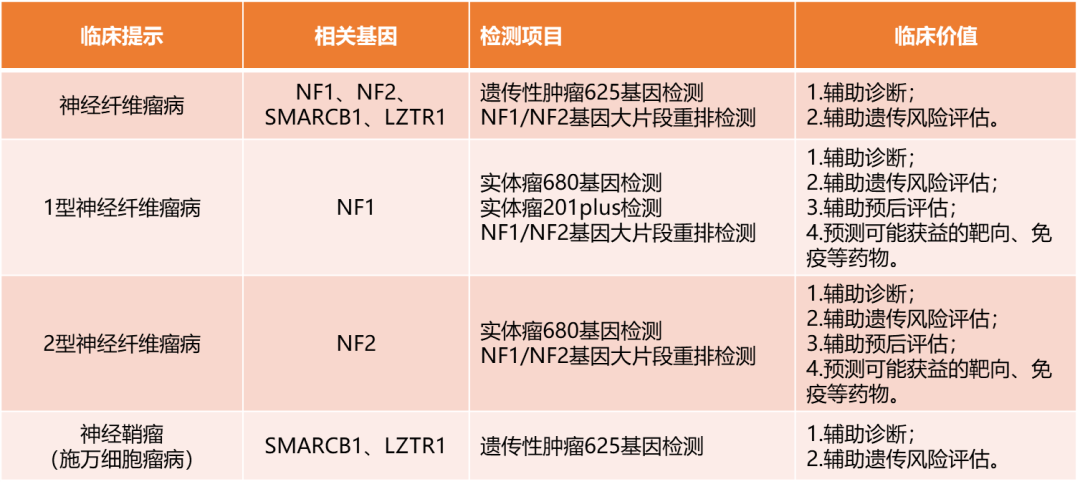
注:NGS项目覆盖了90%以上的NF1和NF2基因变异。若NGS检测结果为阴性,可考虑NF1/NF2基因大片段重排检测。
那么,什么是神经纤维瘤病?基因检测在神经纤维瘤病的精准诊疗中又发挥怎样的作用呢?今天,小编将围绕上述问题,为大家详细解答。
神经纤维瘤病(neurofibromatosis,NF)是一类常染色体显性遗传疾病,在临床和遗传学上有3种主要不同类型:1型神经纤维瘤病(NF1)、2型神经纤维瘤病(NF2)和神经鞘瘤病。该类疾病表型复杂多样,给临床诊疗带来挑战。

图3 3种神经纤维瘤病亚型相关基因变异及其外显率
1型神经纤维瘤病
1型神经纤维瘤病(NF1)是一种由NF1基因突变引起的神经系统常染色体显性遗传疾病,占所有神经纤维瘤病的96%。NF1典型的临床症状包括咖啡牛奶斑(café-au-lait macules,CALMs)、多发性神经纤维瘤、腋窝或腹股沟雀斑等,其中神经纤维瘤是最常见和具有特征性的症状之一。
《儿童及青少年神经纤维瘤病诊疗规范(2021年版)》(以下简称“诊疗规范”)采用2020年公布的新NF1诊断标准。即,6个或以上咖啡牛奶班,在青春期前直径(最长径)>5mm或在青春期后直径>15mm;双侧腋窝或腹股沟区雀斑;2个或以上任何类型的神经纤维瘤或1个丛状神经纤维瘤;2个或以上Lisch结节(虹膜错构瘤),2个或以上的脉络膜异常;视路胶质瘤;特征性骨病变,如蝶骨翼发育不良,胫骨前外侧弯曲(胫骨发育不良)或长骨假关节;NF1基因杂合变异;父母一方通过以上标准被诊断为NF1。满足以上至少2条临床特征,即可诊断为NF1。因NF1涉及全身多部位病变,疑似NF1的儿童应由多学科团队进行评估。
NF1基因位于染色体17q11.2,长度约350kb,包含58个外显子。现已发现1000多种致病性NF1基因变异,包括无义突变、错义突变、剪切突变、大片段重排、染色体微缺失等多种变异类型,且并无热点突变。上海交通大学医学院附属新华医院研究团队分析了109例临床表型为NF1症状的中国患者NF1基因突变情况。检测结果提示,97例患者检出NF1突变,共有89种胚系有害突变。其中21个无义突变,26个移码突变,15个剪切突变,12个错义突变,2例框内缺失,1例缺失,11例染色体微缺失和1例单外显子缺失变异。需要指出的是,染色体微缺失和单外显子缺失变异由MLPA方法检出。

图4 NF1基因的不同外显子区相对突变频率
另一项国外单中心临床研究,入组583例有NF1症状的患者。基于NIH诊断标准,A组患者共365例,满足≥2个临床特征。B组患者218例,只满足1个临床特征。NF1基因突变检测提示,A组患者中,287例检出NF1基因突变,阳性率约为79%,其中87例无义突变,63例错义突变、66例移码突变、38例剪切突变(发生在外显子附近区域)、10例拷贝数缺失以及23例其他突变。B组患者中,64例检出NF1基因突变,阳性率约为29%,其中13例无义突变、22例错义突变、11例移码突变、11例剪切突变、2例拷贝数缺失以及5例其他变异。综上所述,点突变、插入缺失是NF1主要变异形式,拷贝数缺失等其他变异相对少见。
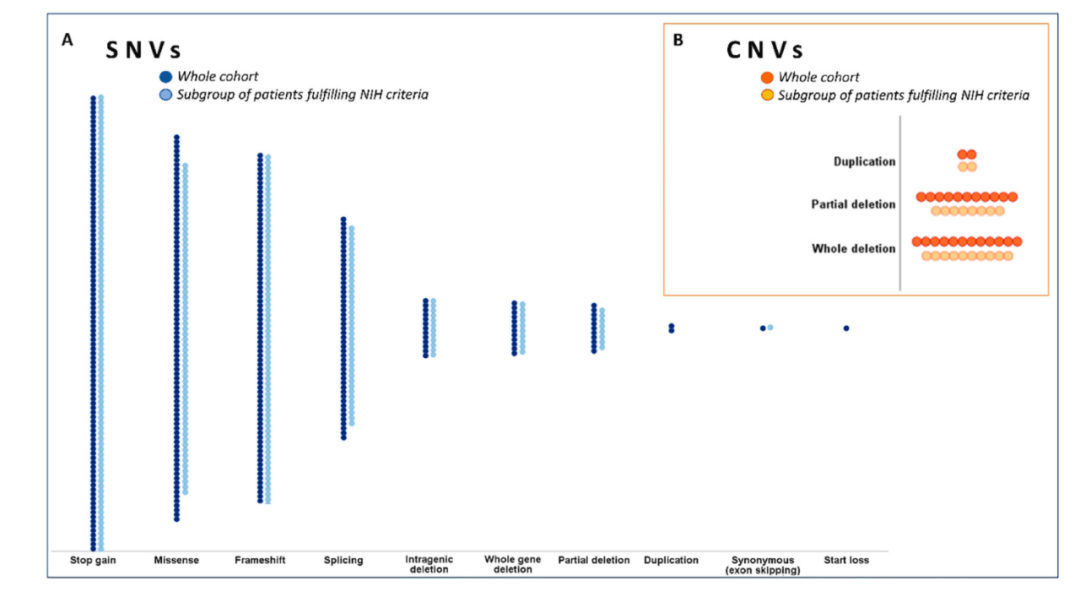
图5 NF1基因不同变异类型分布
《1型神经纤维瘤病临床诊疗专家共识(2021版)》指出,NF1基因不同突变类型、不同突变位点与NF1患者的临床表型之间,存在相关性。目前得到公认的NF1患者基因型-表型相关性如下表所示。发生17号外显子框内缺失突变(c2970-2972delAAT)患者,疾病程度较轻,临床表型为咖啡牛奶斑及雀斑、无神经纤维瘤。而检出NF1基因微缺失患者,疾病程度严重,临床表型为大量神经纤维瘤、智力残疾、心血管畸形、恶性外周神经鞘瘤的风险增加。
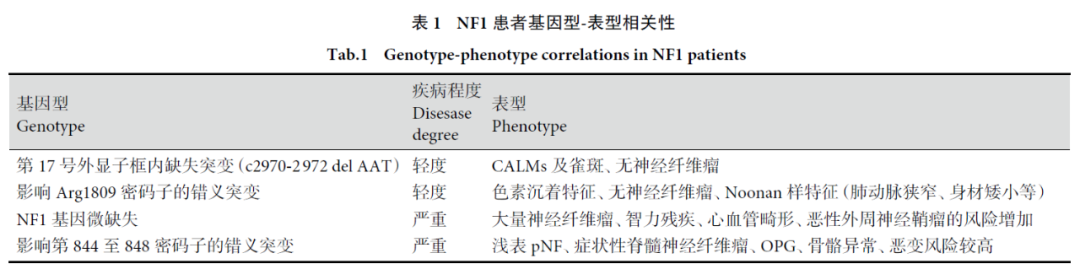
图6 NF1患者基因型—表型相关性
部分1型神经纤维瘤病患者,可考虑靶向药物治疗。若患者临床表型为丛状神经纤维瘤,FDA已经批准司美替尼靶向治疗。若临床表型为视路神经胶质瘤,司美替尼等靶药已处于临床试验中。若临床表型为胃肠道间质瘤,则患者对伊马替尼反应欠佳,舒尼替尼有一定的疗效。
综上所述,结合临床表型,若怀疑为1型神经纤维瘤病,推荐实体瘤201plus或实体瘤680基因检测,覆盖了NF1基因的全部外显子区及毗邻剪切区(±20bp),用于辅助诊断,预后评估,遗传风险评估及靶向药物疗效预测。
2型神经纤维瘤病
2型神经纤维瘤病(NF2)是一种由NF2基因突变引起的神经系统常染色体显性遗传疾病,发病率低于NF1,仅占所有神经纤维瘤病的3%。临床上以中枢神经系统或外周神经系统的多发性肿瘤综合症为特征,中枢神经系统肿瘤包括脑膜瘤、神经鞘瘤、胶质瘤和室管膜瘤等,其中双侧前庭神经鞘瘤为特征性表现。
NF2的主要诊断依据为临床表现。《2型神经纤维瘤病神经系统肿瘤多学科协作诊疗策略中国专家共识》推荐修订后的NIH标准作为NF2的诊断标准,具体如下。
满足A、B、C任意一项确诊条件即可诊断:
诊断条件A:双侧听神经瘤作为独立的诊断条件,可以确诊为NF2。
诊断条件B:不同部位的2个NF2相关肿瘤中,检测到同一NF2基因突变可诊断为NF2。(NF2相关肿瘤包括神经鞘瘤、脑脊膜瘤、室管膜瘤,由于在散发脑膜瘤和神经鞘瘤中亦常可检测出NF2基因突变,因此,必须为同一患者2个不同部位的肿瘤检测出NF2基因发生同一位点的突变,方能诊断NF2)
诊断条件C:满足以下2个主要标准或1个主要标准+2个次要标准可以诊断为NF2。主要标准:单侧听神经瘤;NF2患者的一级亲属;≥2个脑脊膜瘤;在血液或正常组织中检测到NF2基因突变。次要标准a(同类病变可累积计数,如罹患2个神经鞘瘤,则视为满足2个次要标准):室管膜瘤、神经鞘瘤(如主要标准为单侧听神经瘤,则应至少包含1个皮肤神经鞘瘤)。次要标准b(同类病变不可累积计数):青少年囊下或皮质白内障、视网膜错构瘤、40岁以下视网前膜、脑脊膜瘤。
NF2基因位于染色体22q12.2,全长约100kp,包含17个外显子。与NF1基因突变类似,NF2基因也有多种变异类型,例如错义突变、剪切突变、无义突变、移码突变以及大片段缺失。北京天坛医院研究团队获取了15个家系37例NF2患者的样本,其中血液样本31份,组织样本15份(每个家系各1例)。NF2基因检测结果发现,血液和组织样本分别检出15种和18种基因突变,包括2种无义突变、4种错义突变、6种移码突变、1种框内缺失突变和5中剪切位点变异。

图7 37例遗传性2型神经纤维瘤患者NF2基因变异
中国人民解放军总医院研究团队比较分析了12例儿童NF2患者和20例成人NF2患者的基因变异情况。结果表明,12例儿童患者均检测NF2基因胚系变异,其中6例患者携带无义突变、2例框内缺失突变、2例外显子缺失突变、1例剪切突变、1例移码突变。而成人患者主要为NF2剪切突变(10例)。

图8 12例儿童神经纤维瘤2型患者基因突变及其与成人患者的比较
专家共识指出,患者临床表型的轻重程度与NF2基因突变类型存在相关性。NF2基因发生无义或移码突变后会形成截短蛋白,患者的临床表型相对更重,而出现大片段缺失和错义突变患者的表型偏轻。NF2基因突变位点的位置亦与疾病的严重程度有关,如突变发生在3’端(14、15号外显子),则脑膜瘤的数量相对较少、死亡风险更低。整体而言,NF2基因的2-13外显子发生截短突变患者的病情最为严重。

图9 NF2临床症状程度与基因突变的关系
目前,外科切除术与立体定向放射一直是2型神经纤维瘤病相关肿瘤的主要治疗策略,靶向治疗尚处于临床探索中。脑膜瘤诊疗指南推荐检测AKT1,BRAF等基因,提示对应的靶向药物治疗可能获益。
神经鞘瘤病(施万细胞瘤病)
神经鞘瘤病是神经纤维瘤病的第3种病理亚型,约占所有神经纤维瘤病患者的1%,较为罕见。该类患者临床特征为多原发性、非前庭神经的神经鞘瘤,临床表现为疼痛。与NF1和NF2不同,大多数神经鞘瘤病患者为新发病例,家族性发病数量不足15%,遗传给后代几率低。
神经鞘瘤病相关的SMARCB1或LZTR1肿瘤抑癌基因突变,发生在85%的家族性和40%的散发型神经鞘瘤病患者中。由于SMARCB1和LZTR1基因,均位于染色体22q,若发生染色体大片段缺失,也会影响NF2基因。
《神经鞘瘤的诊断、治疗和监测ERN GENTURIS临床实践指南》指出,SMARCB1或LZTR1基因胚系变异可作为神经鞘瘤的诊断依据。携带SMARCB1基因突变的神经鞘瘤患者,很可能导致功能损害性肿瘤,应排除恶性转化风险。LZTR1胚系变异患者罹患单侧前庭神经鞘瘤的风险高。临床上,需要与神经鞘瘤鉴别诊断主要是NF2。根据定义,神经鞘瘤缺乏NF2特征性的双侧前庭神经鞘瘤。

图10 指南指出,SMARCB1或LZTR1基因胚系变异可作为神经鞘瘤病诊断依据
目前,关于神经鞘瘤(施万细胞瘤病)的管理,主要有非手术疼痛管理和手术干预。对于多发性快速增大、肿瘤有疼痛或神经功能缺陷的患者,或者无法手术,可积极考虑贝伐珠单抗治疗。
参考文献:
[1] Radtke, Heather B et al. “Genetic Counseling for Neurofibromatosis 1, Neurofibromatosis 2, and Schwannomatosis-Practice Resource of the National Society of Genetic Counselors.” Journal of genetic counseling vol. 29,5 (2020): 692-714. doi:10.1002/jgc4.1303
[2] Zhang, Jia et al. “Molecular Characterization of NF1 and Neurofibromatosis Type 1 Genotype-Phenotype Correlations in a Chinese Population.” Scientific reports vol. 5 11291. 9 Jun. 2015, doi:10.1038/srep11291
[3] Scala, Marcello et al. “Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study.” Cancers vol. 13,8 1879. 14 Apr. 2021, doi:10.3390/cancers13081879
[4] 王智超, 李青峰. "Ⅰ型神经纤维瘤病临床诊疗专家共识(2021版)." 中国修复重建外科杂志.
[5] 《儿童及青少年神经纤维瘤病诊疗规范(2021年版)》
[6] 中国抗癌协会神经肿瘤专业委员会. "2型神经纤维瘤病神经系统肿瘤多学科协作诊疗策略中国专家共识." 中华神经外科杂志 37.7(2021):6.
[7] 赵赋等. "遗传性2型神经纤维瘤病家系基因突变及临床特点分析." 中华神经外科杂志32.1(2016):5.
[8] 刘羽阳等. "儿童神经纤维瘤病2型基因突变特点和临床特征分析." 中华神经外科杂志 36.5(2020):6.
[9] Evans, D Gareth et al. “ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis.” European journal of human genetics : EJHG vol. 30,7 (2022): 812-817. doi:10.1038/s41431-022-01086-x
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







厉害,谢谢分享
69
厉害
84