
呕吐,不是胃肠炎,不是病毒感染,而是“信号”断了
2024-08-11 梅斯罕见新前沿 MedSci原创 发表于上海

6岁淼淼出现多种症状,经检查确诊为亚历山大病,介绍了该病的病因、分型、诊断及治疗,基因检测显示GFAP基因突变,目前无特效疗法,以对症支持为主。
6岁的淼淼(化名),3年前突然出现呕吐,并逐渐加重,家里人觉得是小毛病,并没有放在心上。令人意外的是,2022年10月份,淼淼开始出现吐字不清,口角向右侧偏斜,左侧眼睑闭合不全。2023年2月他又出现腹痛、进食呛咳。2023年6月开始,淼淼又出现呕吐频繁伴头晕。这几年,淼淼的父母带着他四处求医,但是一直找不到病因。
这次,淼淼父母带着他来到某省一家三甲医院就诊,接诊医生对他进行了神经系统检查,发现孩子的眼球向右侧外展时伴轻微眼震,双下肢近端肌力Ⅳ级,四肢肌张力稍低,双侧膝腱反射亢进,双侧踝阵挛阳性。
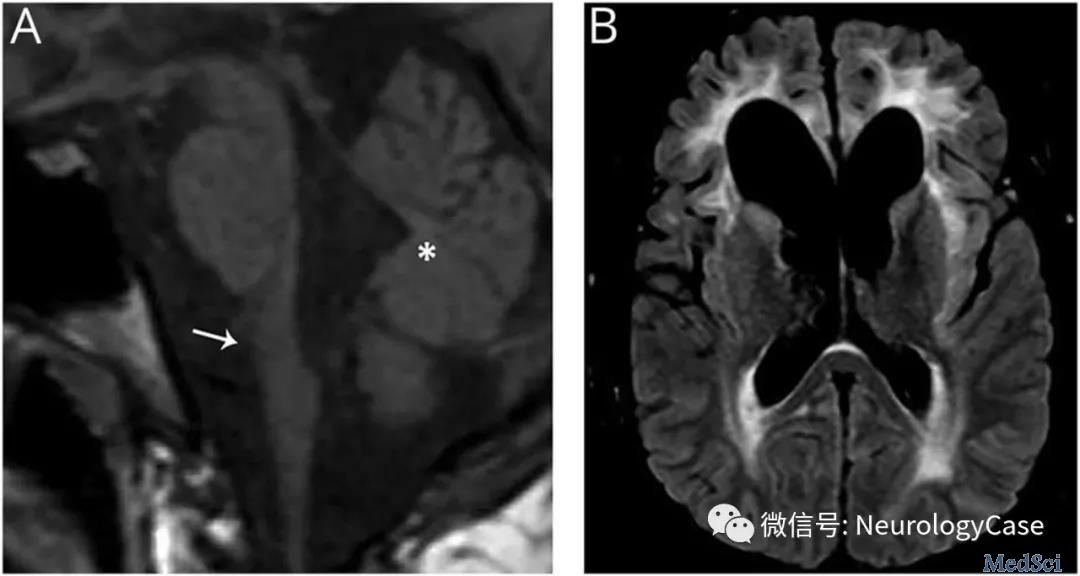
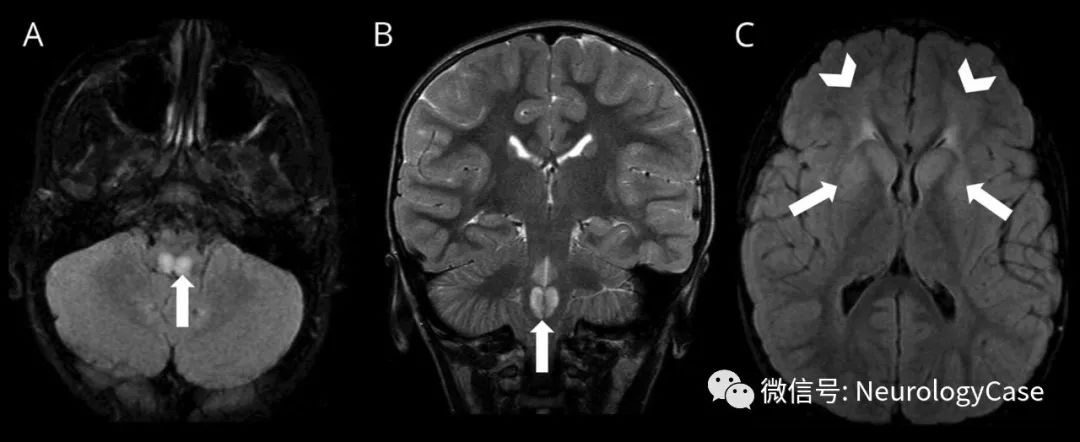
这几年的影像检查结果也不容乐观:头颅MRI平扫(2023年2月6日)提示左侧延髓占位,双侧基底节异常信号。这次的头颅MRI平扫提示双侧脑室旁白质、基底节区可见斑片状稍长T2信号,右侧延髓可见结节样T2信号,Flair呈高信号。脑室系统等大对称,脑沟池裂增宽,脑外间隙略扩大。
淼淼的首发症状为呕吐,随着病程进展逐渐出现吐字不清、饮食呛咳等球麻痹症状,推测为延髓病灶所致,随后患儿出现头晕症状,可能与自主神经功能障碍有关。但是淼淼的临床表现不典型,主要有延髓症状、腭肌阵挛、眼球运动异常、自主神经功能紊乱等。医生怀疑淼淼患有一种罕见病,建议做基因检测。基因检测提示淼淼GFAP基因检测发现外显子1存在c.229A>C(P.N77H)杂合核苷酸变异,符合亚历山大病(Alexander disease,AxD)。经过免疫丙种球蛋白、甲强龙治疗,淼淼的临床症状消失,出院后继续口服泼尼松并逐渐减量治疗。
GFAP基因突变
正常情况下,神经纤维被一层叫做“髓鞘”的脂肪层所覆盖。髓鞘不但能够保护神经纤维,而且帮助它们传递“信号”。亚历山大病(Alexander disease,AxD)是一种致命的神经系统疾病,在亚历山大病患者大脑中,髓鞘被破坏,造成神经“信号”的传递被中断,从而导致神经系统功能受损。
亚历山大病是一种罕见的遗传性脑白质营养不良疾病,多为常染色体显性遗传,主要为编码胶质纤维酸性蛋白的GFAP基因发生突变,进而导致星形胶质细胞功能异常,镜检下星形胶质细胞中存在大量的Rosenthal纤维为其典型的病理特征。多数患有这种疾病的婴儿无法活过1岁。在4-10岁之间患此病的儿童往往会迅速进展。确诊后,患儿可以存活数年,少部分可能活到中年。
分型
根据发病年龄临床上分3型:婴儿型、少年型和成人型。
婴儿型:较其他型多见,起病年龄从生后数月至2岁,多数病例头部缓慢进行性增大,智力低,精神运动发育迟缓,常有抽搐发作,痉挛性瘫痪,可有脑积水,常在幼年死亡,平均病程约为2.5年。
少年型:通常7~14岁起病,主要为进行性智力倒退,甚至痴呆,运动功能障碍,痉挛性截瘫,进行性延髓麻痹,平均病程约为8年左右。
成人型:成年后任何年龄均可起病,部分患者神经系统功能障碍较轻,部分患者间歇性出现神经系统功能障碍。
根据临床症状及MRI图像,将其分为以下三种病型。
大脑优势型(1型):白质脑病,大头症,精神运动发育迟滞,头部MRI:前额部优势的大脑白质病变为特征。主要是婴幼儿期发病,多为功能预后不良的重病例。新生儿期发病例会出现水头症和颅内压亢进症状,生命预后不良。
延髓-脊髓优势型(2型):肌力低下,肌肉痉挛麻痹,球麻痹/假性球麻痹,运动失调,植物性神经障碍等,以核磁共振确认延髓、上位颈髓的信号异常或萎缩为特征。从学童期到成年期以后发病,与其他病型相比,发病过程比较缓慢。
中间型(3型):具有1型和2型两者的特征。发病时期从幼儿期到成年期,范围很广。
诊断
精神症状
1.脑白质病变 2.大头症 3.精神运动发育迟缓 4.四肢运动障碍:肌力低下,痉挛麻痹,小脑性运动失调,肌强直 5.球麻痹或假性球麻痹:吞咽障碍,发音、发声障碍 6.植物神经紊乱:直立性低血压,膀胱直肠障碍,睡眠呼吸暂停 7.腭震颤 8.反复性呕吐
MRI
MRI是诊断亚历山大病有力的工具之一。需要满足以下5个标准中的4个:
(1)前额为主的广泛脑白质病变;
(2)室周边缘存在病变;
(3)基底节和丘脑异常;
(4)脑干异常;
(5)脑室周围区和脑干的病灶强化。
这些诊断标准适用于大多数病例,除了罕见的非典型病例,特别是成人型,表现为极少或无白质病变。成人发病亚历山大病的MRI表现通常与在儿童中观察到的有很大不同,主要表现为脑干下部的萎缩,萎缩可延伸至颈髓。MRI发现的病灶位置通常与大量Rosenthal纤维堆积的区域相关。
基因检查和病理学检查
1.基因检查:确认GFAP基因变异
2.病理学检查:确认大脑白质,上衣下及软膜下的星形细胞质内特征性的:罗氏纤维。
治疗
目前该病没有特效治疗方法,以对症支持治疗为主,由于是遗传性疾病,因此遗传咨询很重要。支持性治疗:包括对一般护理,营养需求,并发感染抗生素治疗以及用于控制癫痫发作的抗癫痫药物(AED)的关注,学习障碍和其他认知障碍的解决方案同没有亚历山大病的人一样。支持性治疗:包括对一般护理,营养需求,并发感染抗生素治疗以及用于控制癫痫发作的抗癫痫药物(AED)的关注,学习障碍和其他认知障碍的解决方案同没有亚历山大病的人一样。
参考资料:
1. Yoshida, T., [Clinical characteristics and diagnostic criteria on Alexander disease]. Rinsho Shinkeigaku, 2020. 60(9): p. 581-588.
2. Messing, A., Alexander disease. Handb Clin Neurol, 2018. 148: p. 693-700.
3. Tavasoli, A., et al., Alexander Disease. Journal of Child Neurology, 2017. 32(2): p. 184-187.
4. van der Knaap, M.S., et al., Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol, 2001. 22(3): p. 541-52.
5. Gopal, N., et al., Teaching NeuroImages: Neuroimaging in adult-onset Alexander disease. Neurology, 2020: p. 10.1212/WNL.0000000000010803.
6. 王秋菊, 沈亦平, 邬玲仟,等. 遗传变异分类标准与指南[J]. 中国科学:生命科学, 2017(6).王秋菊, 沈亦平, 邬玲仟,等. 遗传变异分类标准与指南[J]. 中国科学:生命科学, 2017(6).
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#亚历山大病# #GFAP基因#
48