第十三届江西省心血管病高峰论坛:肥厚型心肌病管理新进展
2022-12-16 MedSci原创 MedSci原创 发表于上海

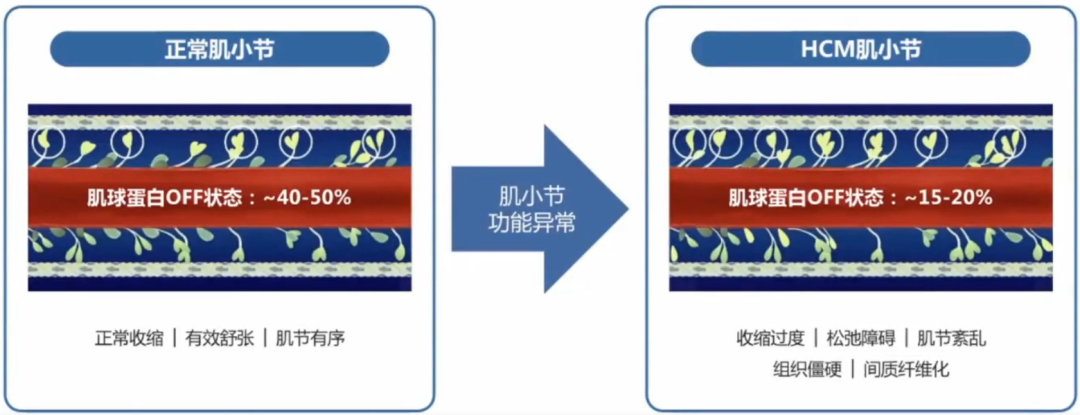
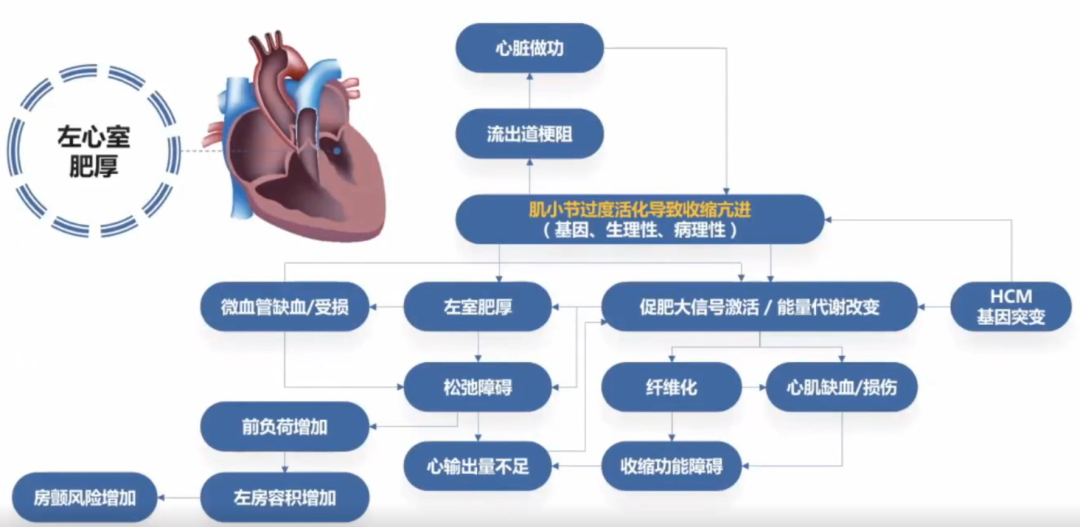
肥厚型心肌病的病因尚未完全明确,编码心肌肌小节相关蛋白的基因突变是主要原因。以肌小节功能异常为核心,肥厚型心肌病的病理生理机制逐渐明确。
2011ACCF/AHA指南:HCM是一种不明原因的以左室肥厚为特征的疾病,不伴有心室腔扩大,除外其他引起左室肥厚的心血管或全身疾病。
2014ESC指南:HCM是指并非完全因心脏负荷异常引起的左心室壁增厚。
2017中国指南:HCM是一种以心肌肥厚为特征的心脏疾病,主耍表现为左心室壁增厚。需排除负荷增加如高血压、主动脉瓣狭窄和先天性主动脉瓣下隔膜等引起的左心室壁增厚。
2020AHA指南:HCM是一类由于跳小节蛋白编码基因(或肌小结蛋白相关基因)变异,或遗传病因不明的以左室心肌肥厚为特征的心脏疾病。排除有明确证据证实其他心脏、系统性或代谢性疾病导致左室肥厚的情况。
2022中国指南:HCM是一种呈常染色体显性遗传的原发性心肌病。主要编码心肌肌小结相关蛋白的基因致病性变异引起,临床表现以心室壁增厚为突出特征。需除外其他可引起心室壁增厚的生理因素、心脏疾病、系统性疾病或代谢性疾病。
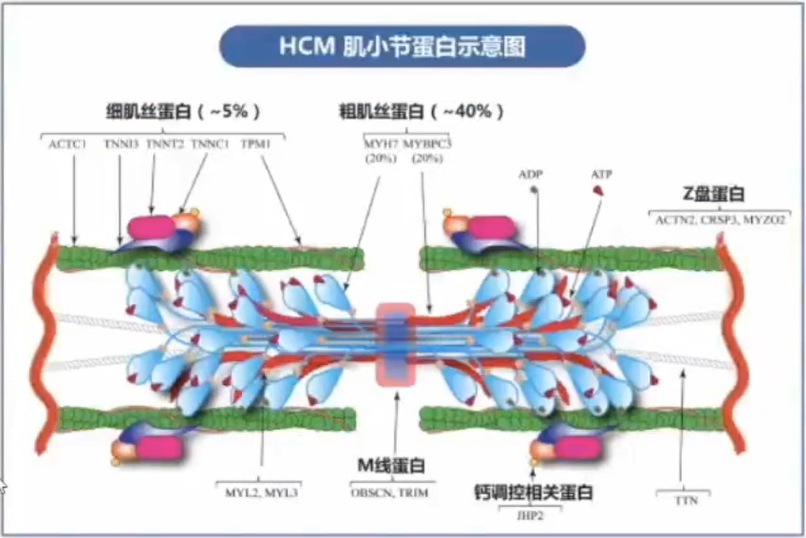
多数HCM是一种单基因遗传性心脏病,为常染色体显性遗传模式。大约60%的HCM患者存在致病基因变异,主要编码心肌肌小节相关蛋白,包括粗肌丝、中间丝及细肌丝等。迄今已在至少8个编码心肌肌小节相关蛋白的基因中发现>1500个与HCM发病相关的变异。MYBPC3基因和MYH7基因是HCM患者最常见的两种致病基因,二者约占基因变异阳性患者的70%。
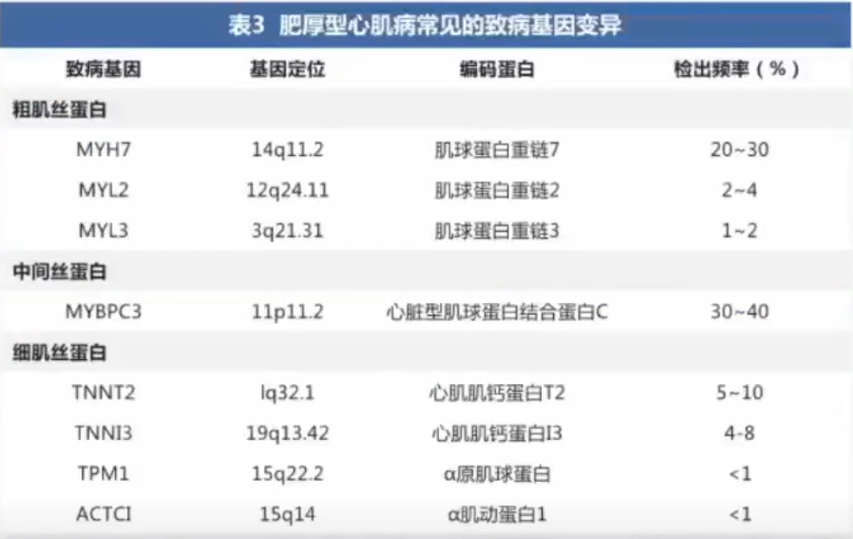
HCM从基因突变到临床表现尚有许多未知。
由于致病基因变异存在外显不全及与年龄依赖的表达,携带致病基因变异不一定出现临床表型。
部分HCM患者可能携带单个基因的多个变异(复合变异)或≥2个相同或不同基因的杂合变异,统称为复杂基因变异现象。研究发现,携带复杂基因变异者发病更早,临床表型更重,预后更差。
仍有高达40%的HCM患者未检测到致病基因变异,主要见于一些散发病例(称为“非家族性HCM”)或小型家系,通常发病较晚,临床表型相对较轻,提示可能有不同机制参与HCM发病。
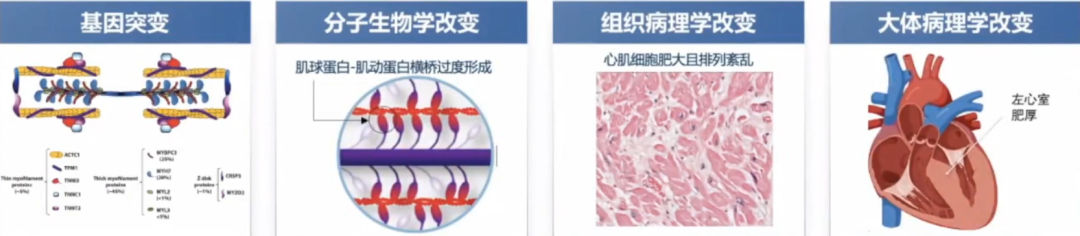
对肌球蛋白这一分子马达的研究,促进了对HCM的认识。
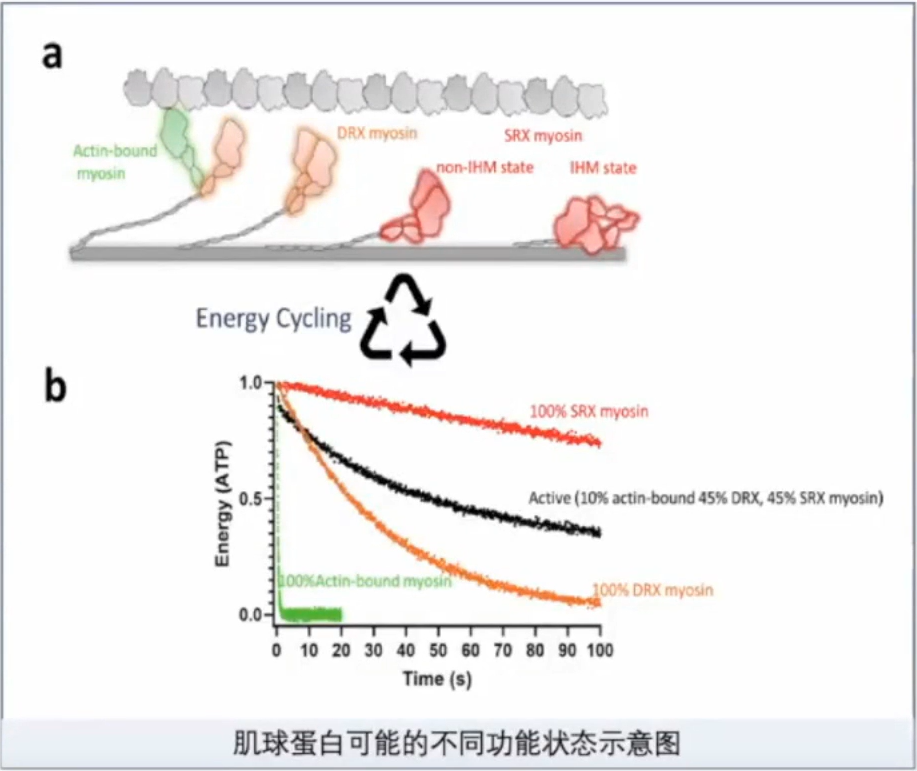
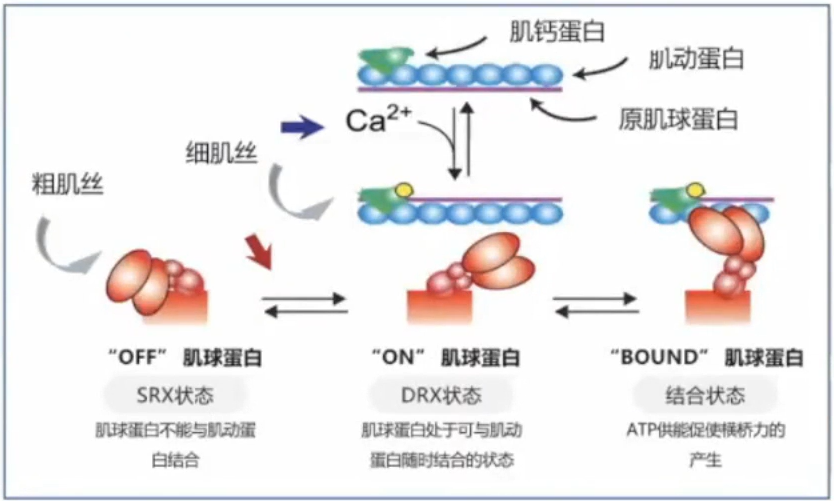
DRX(disordered relaxed):肌球蛋白头处于可以自由与肌动蛋白结合的状态,ATP转换时间小于30s。
SRX(super-relaxed):肌球蛋白头处于不能与肌动蛋白结合的状态,ATP转换时间大于100s。
DRX-SRX的平衡对肌球蛋白和肌动蛋白横桥的形成产生影晌。
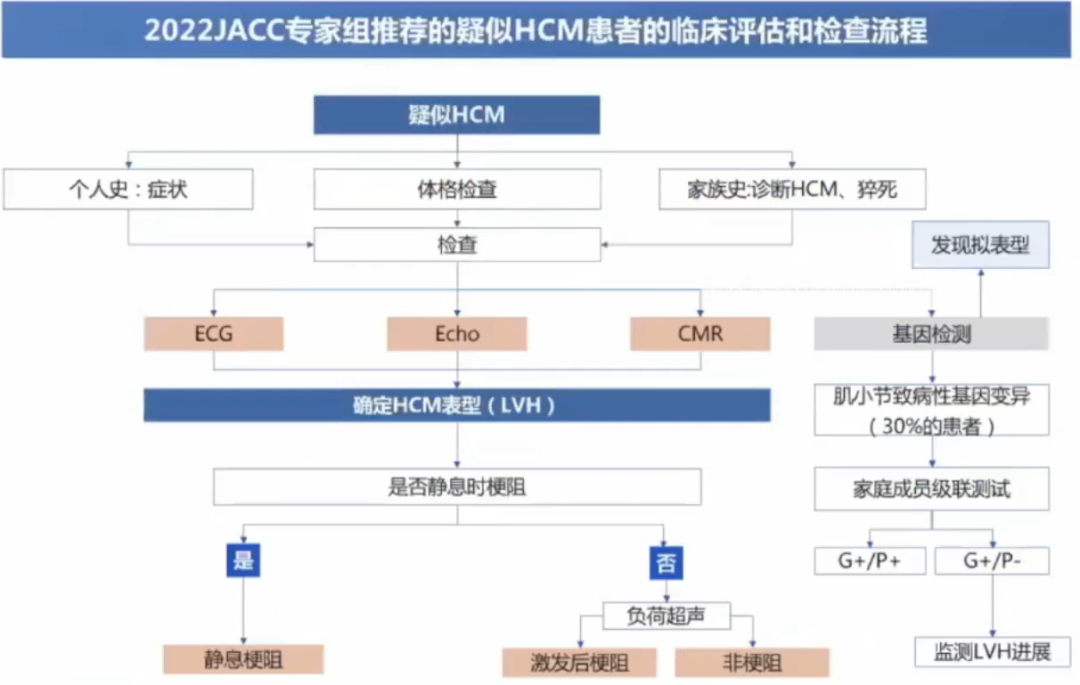
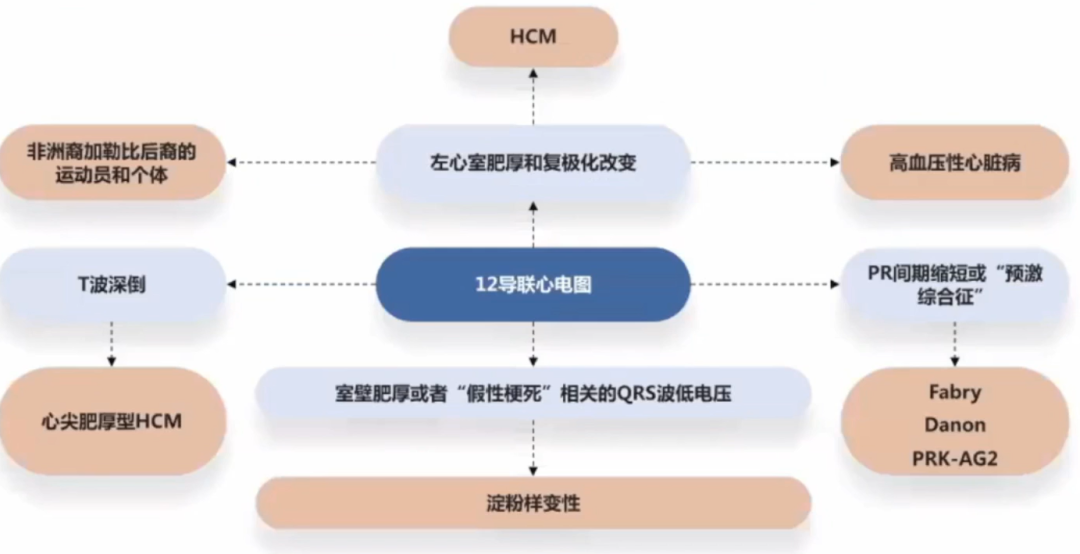
心电图对HCM可提供早期诊断、鉴别诊断的线索。

超声心动图:经胸超声心动图是大部分HCM患者的首选检查方法,用于疾病诊断、确定肥厚程度、筛查左室心尖室壁瘤、评估左室收缩和舒张功能、二尖瓣功能及筛查评估左室流出道梗阻严重程度。
心脏磁共振:CMR可以识别左室心肌肥厚的重点区城,准确评测最大室壁厚度,且可用于评估心肌纤维化程度。在诊断不明确、超声成像不佳、无法明确是否植入ICD时,CMR可作为有效的辅助检查。
冠脉CTA:可用于无创评估HCM患者的冠状动脉。
核素:心肌灌注显像可以发现缺血和评估严重程度,PET优于SPECT。
基因检测是鉴别拟表型和家系评估的重要手段
首选包含8个与HCM发病明确相关的“核心致病基因”(MYH7、MYBPC3、MYL2、MYL3、TNNT2、TNNI3、TPM1和ACTCl基因)的“靶基因”或“基因组合”测序(I类推荐,B级证据)。
如果上述基因检测未能明确致病基因突变,可以考虑扩展为针对心肌病的基因组合测序,或全外显子组测序或全基因组测序。
如果考虑其他HCM“拟表型”时,推荐进行相关特定基因突变检测(I类推荐,B级证据)。
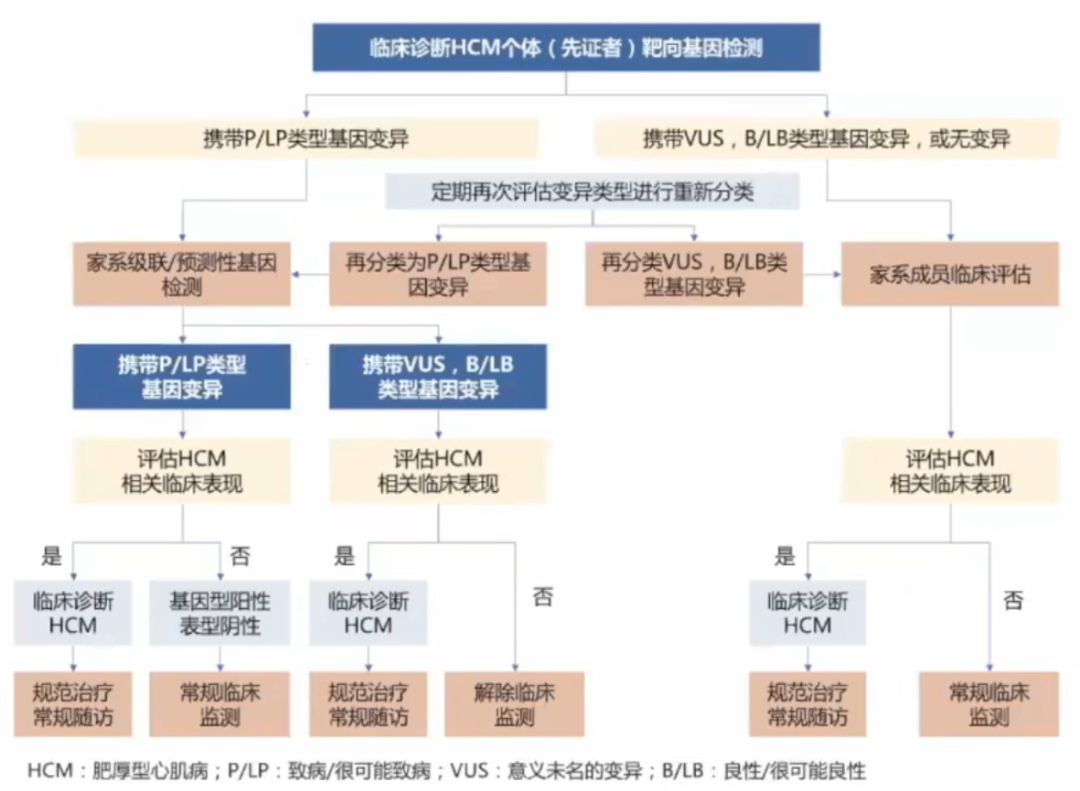
基因变异检测结果只有致病(Pathological,P)和很可能致病(likely pethological,LP)类型的变异才是有临床意义的。
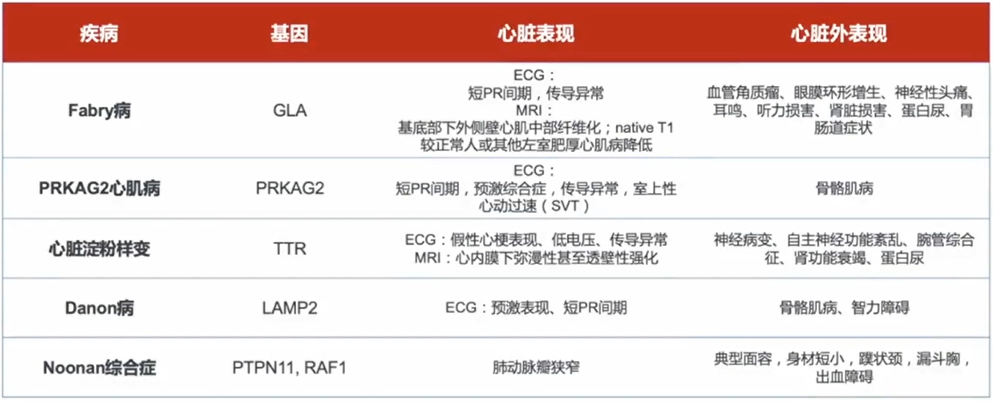
运动员心脏改变:无HCM家族史,心肺运功功能较好,通常不合并左心房增大。
高血压性心肌肥厚:高血压病史较长,心肌肥厚通常呈对称性。
主动脉瓣狭窄:左心室和室间隔呈对称性肥厚,主动脉根部狭窄后扩张。
肥厚型心肌病:不可溶性淀粉样前蛋白沉积于组织或器官的细胞外区,导致结构和功能障碍,心脏是淀粉样变常累积的器官,表现为心肌肥厚和舒张受限。
心肌淀粉样变:不可溶性淀粉样前蛋白沉积于组织或器官的细胞外区,导致结构和功能障碍,心脏是淀粉样变常累积的器官,表现为心肌肥厚和舒张受限。
与其他代谢疾病、神经肌肉疾病鉴别:如法布雷病、Danon病、Pompe病、Friedreich共济失调、线粒体心肌病等(有HCM表型的临床综合征)。
提高HCM拟表型的鉴别诊断:
HCM疾病管理是一项复杂的工作,需要包含:心脏评估、家系筛查、药物治疗和/或SRTs、SCD危险分层及预防、心律失常管理、生活方式干预。
驰缓肥厚心肌,逆转心肌肥厚,延长寿命;抗心律失常,维持正常窦律,预防猝死;防治血栓栓塞事件;减轻左室流出道狭窄,缓解症状;避免使用增加心肌收缩力和减少心脏容量的药物:洋地黄类药(除非发生快速室上性心律失常或收缩功能不全),硝酸类制剂,利尿剂慎用;生活指导,避免激烈运动、屏气、持重。
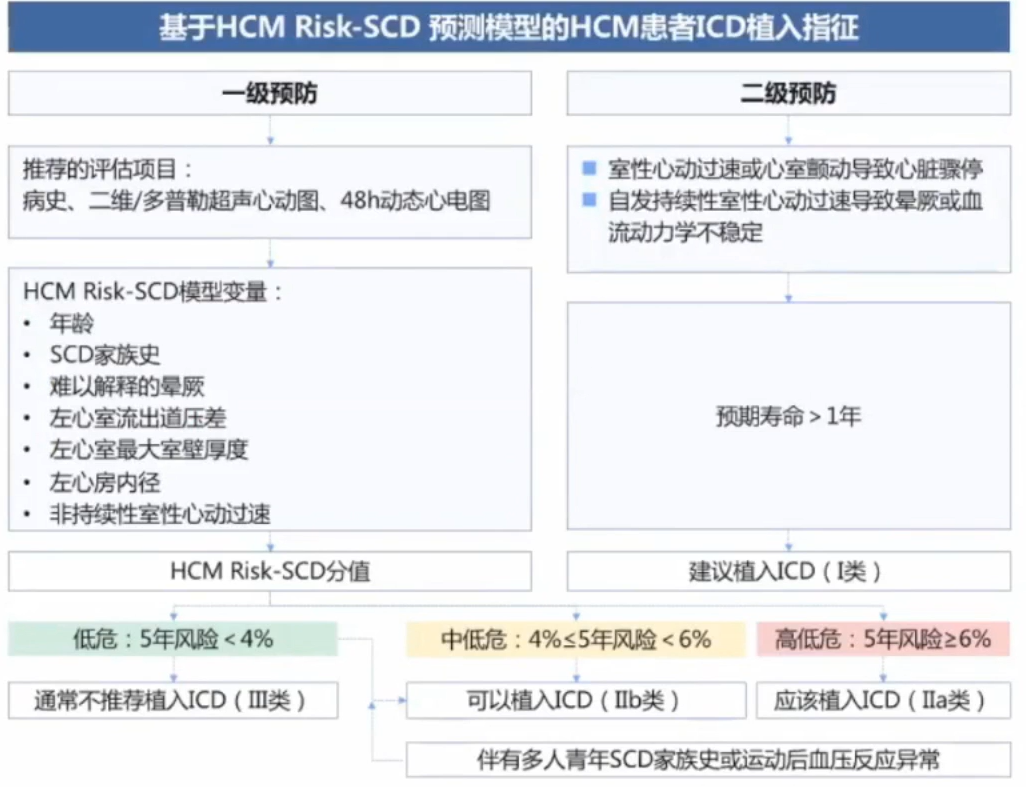
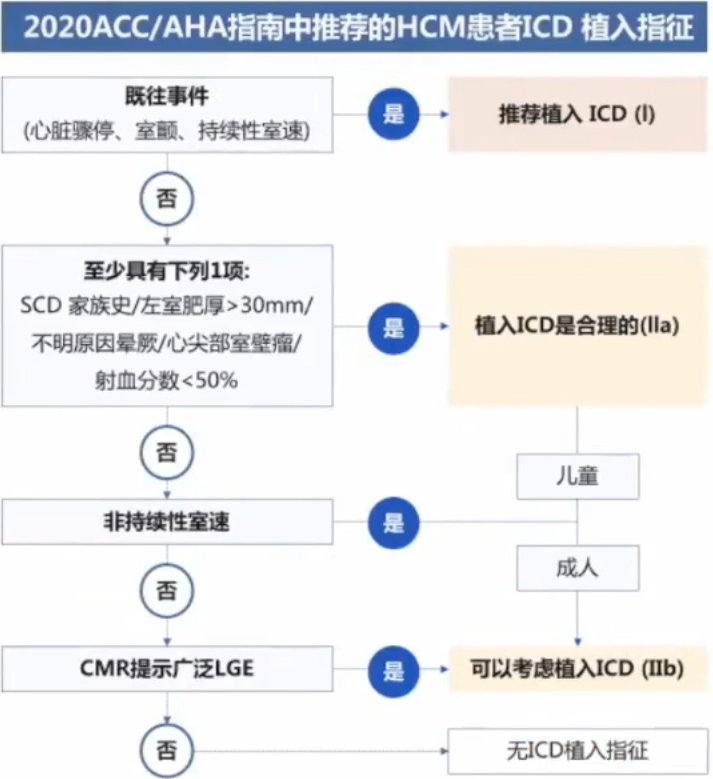
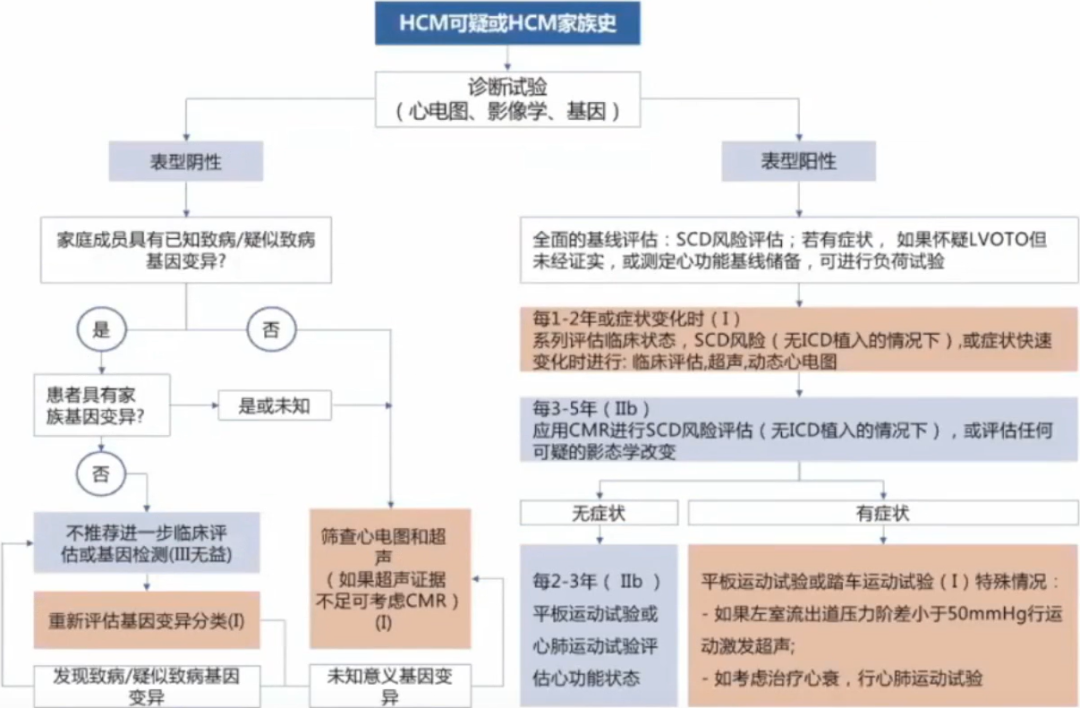
HCM是一种高度异质性的心脏疾病,从婴儿到老年人均有可能发病。
大多数HCM患者需要密切随访。如果一级亲属携带与先证者相同的致病基因变异但不具有LVH的证据且没有症状,诊断为“基因型阳性表型阴性”个体,推荐长期定期随访,监测疾病的进展。
临床评估内容包括症状评估和辅助检查,症状评估主要指活动相关的呼吸困难、胸痛、心悸或晕厥等,辅助检查至少包括12导联心电图及超声心动图检查。如果亲属未检测到致病基因变异,初次临床评估也无HCM相关的临床表现,则可以解除进一步的临床监测。
HCM目前的治疗手段有限。
HCM药物治疗一直无突破性进展,现有药物并不能充分改善患者症状或者耐受性较差。当前可用的药物不具有疾病特异性,不能解决HCM发病的核心分子机制-肌小结病变。
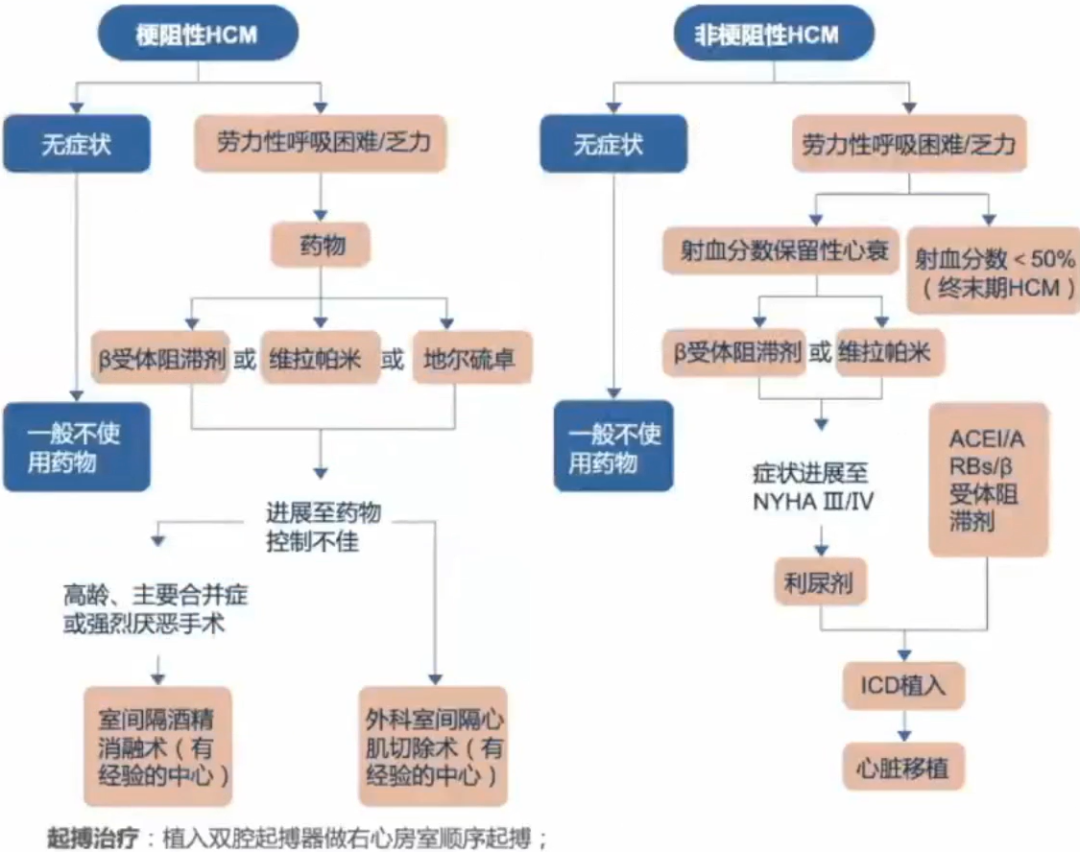
新的手术方式:外科乳头肌重建术、脊索切除、二尖瓣修复、心尖肌切除术、经导管二尖瓣修补术、射频消融术、高强度聚焦超声间隔消融术。
新的药物治疗:Mavacamten、CK-274、派克昔林、曲美他嗪、雷诺嗪、Eleclazine、N-乙酰半胱氨酸、血管紧张素受体阻滞剂、醛固酮拮抗剂、他汀类。
基因疗法:等位基因特异性基因沉默,使用CRISPR/Cas9修复胚胎基因。
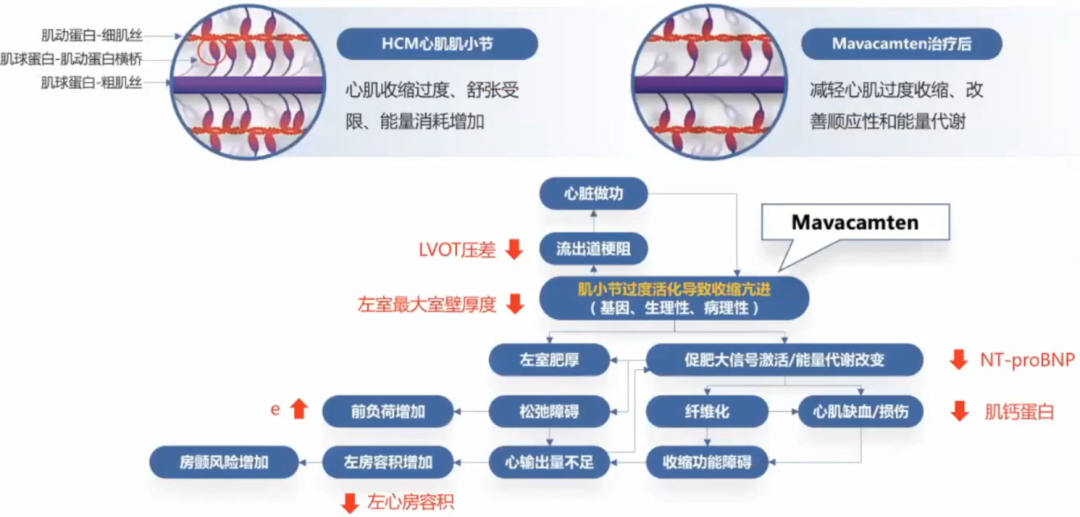
靶向肥厚型心肌病核心病理生理机制的心肌肌球蛋白抑制剂,完成从实验室到临床之路。


肥厚型心肌病的病因尚未完全明确,编码心肌肌小节相关蛋白的基因突变是主要原因。以肌小节功能异常为核心,肥厚型心肌病的病理生理机制逐渐明确。
肥厚型心肌病临床症状的非特异性和疾病表现的异质性给临床诊疗带来挑战,临床评估应结合症状,合理运用心电图、超声、磁共振等手段,同时考虑基因检测在鉴别拟表型和家系评估中的重要作用。
肥厚型心肌病目前的治疗方法针对症状缓解。新的治疗方法包括新的手术方法、新的药物靶点以及基因治疗在不断探索。心肌肌球蛋白抑制剂Mavacamten,通过改善肌小节功能,改善HCM病理生理通路多个环节,获得临床验证,逐步接近肥厚型心肌病的治疗目标。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言